一种化合物及其制备方法与应用与流程
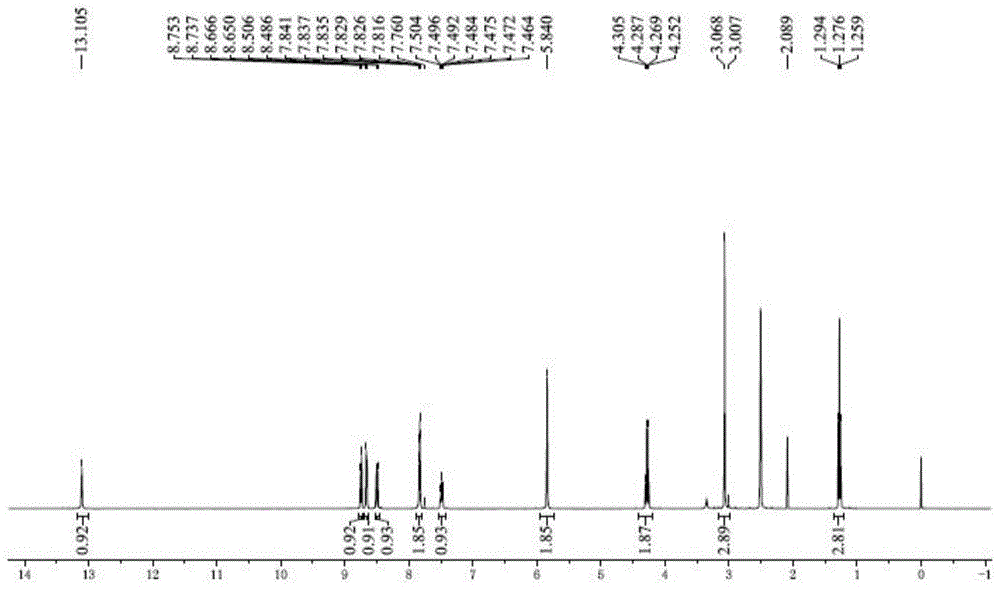
本发明涉及抗癌与止痛药物领域,尤其涉及一种化合物及其制备方法与应用。
背景技术:
:癌症已经成为当今世界的一大杀手,给社会和患者家庭造成巨大的负担。癌症的化学药物治疗(化疗)是常规性的治疗措施,但化疗药物往往毒性大,造成患者诸如肝肾功能损伤与骨髓抑制等副作用,因而医药学家一直在寻求有效且毒性小的抗癌药物。疼痛是癌症患者的常见症状。充分的疼痛评估和疼痛管理对于提高该人群的生活质量和健康结果至关重要。全球最高影响因子期刊acancerjournalforclinicians,ca(if=224)。同时刊登了两篇文章:“optimalpainmanagementforpatientswithcancerinthemodernera”及“painmanagementforpatientswithcancer”。都同时指出:缓解疼痛在治疗癌症中是十分有必要的。在以前的文献中,64%的晚期或转移性癌症患者报告疼痛;目前59%接受抗癌治疗的患者报告疼痛,1/3的患者在完成治疗后仍有疼痛感。临床缺乏疼痛管理专家、不愿意开阿片类药物以及被认为存在的监管限制常常被认为是障碍,但最普遍的障碍之一是提供者对疼痛管理知识不足。在以往的疼痛管理中一般为以下步骤:第一步,使用非处方止痛药来管理疼痛;第二步,逐步升级到使用传统被认为的弱阿片类药物;第三步,提倡使用更强的阿片类药物;第四步,提醒临床医生考虑对疼痛的非药物治疗方案使用干预措施;干预措施为硬膜外或鞘内镇痛药和神经阻滞。一般可使用多种药物的组合物来帮助控制疼痛,这些药物包括:对乙酰氨基酚、阿司匹林、阿片类药物以及其他常用于治疗其他疾病(如抑郁症或癫痫症)的药物。虽然这些多种药物的组合比一种药物效果更好,但是长时间服用,易引起过敏反应、胃肠道反应、肝、肾、肺损害、心力衰竭等一系列综合征。综上所述,寻求有效的抗癌药物和进行癌症疼痛的治疗与管理非常重要,但是现有技术中尚无一种既有明确的抗癌效果又能同时有效缓解疼痛的药物。技术实现要素:本发明的目的在于克服现有技术中的缺陷,提供一种化合物并应用在制备治疗各类癌症及疼痛药物中的应用。为了实现上述发明目的,本发明提供以下技术方案:本发明提供了2-(2-乙氧基-2-氧乙基)-1-甲基-9h-吡啶并[3,4-b]吲哚-2-溴化铵,具有式1所示结构,本发明提供了所述2-(2-乙氧基-2-氧乙基)-1-甲基-9h-吡啶并[3,4-b]吲哚-2-溴化铵的制备方法,包含以下步骤:(1)将l-色氨酸、乙醛和乙酸混合后进行加成反应,得到3s-1-甲基-2,3,4,9-四氢-吡啶并[3,4-b]吲哚-3-羧酸;所述加成反应的反应过程如下:(2)将所述的3s-1-甲基-2,3,4,9-四氢-吡啶并[3,4-b]吲哚-3-羧酸、n,n-二乙基乙胺、二甲基甲酰胺和n-氯代丁二酰亚胺混合后进行氧化反应,得到1-甲基-9h-吡啶并[3,4-b]吲哚;所述氧化反应的反应过程如下:(3)将所述的1-甲基-9h-吡啶并[3,4-b]吲哚、溴乙酸乙酯和丙酮混合后进行反应,得到2-(2-乙氧基-2-氧乙基)-1-甲基-9h-吡啶并[3,4-b]吲哚-2-溴化铵;所述反应的反应过程如下:作为优选,所述步骤(1)中的l-色氨酸、乙醛和乙酸的用量比为1g:(20~28)g:(3~7)ml。作为优选,所述步骤(1)中加成反应的反应温度为70~90℃,所述加成反应的时间为0.5~1.5h。作为优选,所述步骤(2)中的3s-1-甲基-2,3,4,9-四氢-吡啶并[3,4-b]吲哚-3-羧酸、n,n-二乙基乙胺、二甲基甲酰胺和n-氯代丁二酰亚胺的用量比为(3~4)g:(5~10)g:(15~19)ml:(10~14)g。作为优选,所述步骤(2)中氧化反应的反应温度为20~30℃,所述氧化反应的反应时间为10~50min。作为优选,所述步骤(3)中的1-甲基-9h-吡啶并[3,4-b]吲哚、溴乙酸乙酯和丙酮的用量比为1g:(1.3~1.5)g:(7.2~12.9)ml。作为优选,所述步骤(3)中反应的反应温度为50~70℃,所述反应的反应时间为14~18h。本发明还提供了一种由2-(2-乙氧基-2-氧乙基)-1-甲基-9h-吡啶并[3,4-b]吲哚-2-溴化铵制备得到的化合物,具有式2所示结构,本发明还提供了式2所述化合物的制备方法,反应步骤如下:将2-(2-乙氧基-2-氧乙基)-1-甲基-9h-吡啶并[3,4-b]吲哚-2-溴化铵、1,2-环己二酮、甲醇钠和二甲基甲酰胺混合后,反应温度为90~110℃,反应时间为14~18h,得到2,3,4,13-四氢-1h-吲哚并[2',3':3,4]吡啶并[1,2-b]异喹啉-6-溴化铵;所述反应的反应过程如下:作为优选,所述2-(2-乙氧基-2-氧乙基)-1-甲基-9h-吡啶并[3,4-b]吲哚-2-溴化铵、1,2-环己二酮、甲醇钠和二甲基甲酰胺的用量比为1g:(0.3~0.5)g:(0.5~0.7)g:(13~15)ml。本发明还提供了一种由2-(2-乙氧基-2-氧乙基)-1-甲基-9h-吡啶并[3,4-b]吲哚-2-溴化铵制备得到的化合物,具有式3所示结构,本发明还提供了式3所述化合物的制备方法,反应步骤如下:将2-(2-乙氧基-2-氧乙基)-1-甲基-9h-吡啶并[3,4-b]吲哚-2-溴化铵、1,2-环己二酮、甲醇钠和二甲基甲酰胺混合后,反应温度为50~70℃,反应时间为0.5~1.5h,得到5-羧基-2,3,4,13-四氢-1h-吲哚并[2',3':3,4]吡啶并[1,2-b]异喹啉-6-溴化铵;所述反应的反应过程如下:作为优选,所述2-(2-乙氧基-2-氧乙基)-1-甲基-9h-吡啶并[3,4-b]吲哚-2-溴化铵、1,2-环己二酮、甲醇钠和二甲基甲酰胺的用量比为1g:(400~430)mg:(580~620)mg:(14~24)ml。本发明还提供了式1、式2或式3任意一种化合物在制备治疗癌症疼痛药物中的应用。本发明提供了可用于制备癌症止痛药的化合物。本发明还提供了所述化合物的制备方法。本发明提供的合成方法操作简单,具有普适性,适于批量生产。本发明提供所述化合物在制备治疗癌症疼痛药物中的应用。通过本发明所述化合物制备得到的药物,可有效的降低癌症治疗中对阿片类药物的依赖,并建立良好的疼痛管理机制。附图说明图1为实施例1中2-(2-乙氧基-2-氧乙基)-1-甲基-9h-吡啶并[3,4-b]吲哚-2-溴化铵的核磁共振氢谱图;图2为实施例1中2,3,4,13-四氢-1h-吲哚并[2',3':3,4]吡啶并[1,2-b]异喹啉-6-溴化铵的核磁共振氢谱图;图3为生物碱a对神经胶质瘤(u251)细胞移植瘤生长抑制的表型图;图4为生物碱a处理后的斑马鱼神经胶质瘤(u251)细胞移植瘤荧光强度;图5为生物碱a对神经胶质瘤(u251)细胞移植瘤的生长抑制作用;图6生物碱a对正常斑马鱼幼鱼的存活率的影响;图7各组斑马鱼给药前后自身荧光强度降低率。具体实施方式本发明提供了2-(2-乙氧基-2-氧乙基)-1-甲基-9h-吡啶并[3,4-b]吲哚-2-溴化铵,具有式1所示结构,本发明提供了所述2-(2-乙氧基-2-氧乙基)-1-甲基-9h-吡啶并[3,4-b]吲哚-2-溴化铵的制备方法,包含以下步骤:(1)将l-色氨酸、乙醛和乙酸混合后进行加成反应,得到3s-1-甲基-2,3,4,9-四氢-吡啶并[3,4-b]吲哚-3-羧酸;所述加成反应的反应过程如下:(2)将所述的3s-1-甲基-2,3,4,9-四氢-吡啶并[3,4-b]吲哚-3-羧酸、n,n-二乙基乙胺、二甲基甲酰胺和n-氯代丁二酰亚胺混合后进行氧化反应,得到1-甲基-9h-吡啶并[3,4-b]吲哚;所述氧化反应的反应过程如下:(3)将所述的1-甲基-9h-吡啶并[3,4-b]吲哚、溴乙酸乙酯和丙酮混合后进行反应,得到2-(2-乙氧基-2-氧乙基)-1-甲基-9h-吡啶并[3,4-b]吲哚-2-溴化铵;所述反应的反应过程如下:本发明将l-色氨酸、乙醛和乙酸混合后进行加成反应,得到3s-1-甲基-2,3,4,9-四氢-吡啶并[3,4-b]吲哚-3-羧酸。在本发明中,所述步骤(1)中的l-色氨酸、乙醛和乙酸的用量比优选为1g:(20~28)g:(3~7)ml,更优选为1g:(22~26)g:(4~6)ml,进一步优选为1g:(24~25)g:(4.5~5.5)ml。在本发明中,所述步骤(1)中加成反应的反应温度优选为70~90℃,进一步优选为75~85℃;所述加成反应的时间优选为0.5~1.5h,进一步优选为0.75~1.25h。本发明所述加成反应过程中,l-色氨酸和乙醛作为反应原料,在乙酸溶剂中加成得到3s-1-甲基-2,3,4,9-四氢-吡啶并[3,4-b]吲哚-3-羧酸。在本发明中,所述步骤(1)中加成反应结束后,优选对得到的产物体系依次进行减压浓缩、稀释、调节ph、洗涤,过滤后所得的淡黄色固体即为3s-1-甲基-2,3,4,9-四氢-吡啶并[3,4-b]吲哚-3-羧酸。在本发明中,所述步骤(1)中l-色氨酸与稀释用水的质量比优选为1:(3~6),更优选为1:(4~5);所述调节ph的试剂优选为氨水;所述调节ph的ph值优选为7;所述洗涤的洗涤顺序优选为水和乙腈的依次洗涤;所述l-色氨酸与洗涤用水的质量比优选为1:(3~6),更优选为1:(4~5);所述l-色氨酸与乙腈的用量比优选为1g:(1~3)ml,更优选为1g:(1.75~2.25)ml。得到3s-1-甲基-2,3,4,9-四氢-吡啶并[3,4-b]吲哚-3-羧酸后,本发明将3s-1-甲基-2,3,4,9-四氢-吡啶并[3,4-b]吲哚-3-羧酸、n,n-二乙基乙胺、二甲基甲酰胺和n-氯代丁二酰亚胺混合后进行氧化反应,得到1-甲基-9h-吡啶并[3,4-b]吲哚。在本发明中,所述步骤(2)中的3s-1-甲基-2,3,4,9-四氢-吡啶并[3,4-b]吲哚-3-羧酸、n,n-二乙基乙胺、二甲基甲酰胺和n-氯代丁二酰亚胺的用量比优选为(3~4)g:(5~10)g:(15~19)ml:(10~14)g,进一步优选为(3.25~3.75)g:(6~9)g:(16~18)ml:(11~13)g,更优选为(3.45~3.55)g:(7~8)g:(16.5~17.5)ml:(11.5~12.5)g。在本发明中,所述混合优选在0℃下进行。在本发明中,所述步骤(2)中氧化反应的反应温度优选为20~30℃,进一步优选为22.5~27.5℃;所述氧化反应的反应时间优选为10~50min,进一步优选为20~40min,更优选为25~35min。在本发明中,所述氧化反应的反应时间到达以后,本发明优选加入淬灭试剂淬灭所述氧化反应。在本发明中,所述步骤(2)中淬灭反应的淬灭试剂优选为水;所述n,n-二乙基乙胺与淬灭试剂的用量比优选为1g:(6~7.38)ml,进一步优选为1g:(6.5~6.88)ml。在本发明中,所述步骤(2)中氧化反应的反应物优选为n,n-二乙基乙胺和n-氯代丁二酰亚胺,所述步骤(2)中氧化反应的溶剂优选为二甲基甲酰胺。在本发明中,所述步骤(2)中氧化反应结束后,优选对得到的产物体系依次进行萃取、洗涤1、干燥、减压过滤、洗涤2、过滤后所得的黄色固体即为1-甲基-9h-吡啶并[3,4-b]吲哚。在本发明中,所述萃取所用的有机溶剂优选为乙酸乙酯,所述n,n-二乙基乙胺与乙酸乙酯的用量比优选为1g:(15~25)ml,进一步优选为1g:(17~23)ml;所述洗涤1优选为饱和碳酸钠溶液和盐水的依次洗涤,所述n,n-二乙基乙胺与饱和碳酸钠溶液的用量比优选为1g:(15~25)ml,进一步优选为1g:(17~23)ml,所述盐水与饱和碳酸钠溶液的体积比为1:(3.5~4),进一步优选为1:(3.6~3.9);所述干燥所用的试剂优选为硫酸钠;所述洗涤2所用的试剂优选为乙醚,所述乙醚与饱和碳酸钠溶液的体积比优选为1:(4~8),进一步优选为1:(5~7)。得到1-甲基-9h-吡啶并[3,4-b]吲哚后,本发明将1-甲基-9h-吡啶并[3,4-b]吲哚、溴乙酸乙酯和丙酮混合后进行反应,得到2-(2-乙氧基-2-氧乙基)-1-甲基-9h-吡啶并[3,4-b]吲哚-2-溴化铵。在本发明中,所述步骤(3)中的1-甲基-9h-吡啶并[3,4-b]吲哚、溴乙酸乙酯和丙酮的用量比优选为1g:(1.3~1.5)g:(7.2~12.9)ml,进一步优选为1g:(1.35~1.45)g:(9~10)ml。在本发明中,所述步骤(3)中反应的反应温度优选为50~70℃,进一步优选为55~65℃;所述反应的反应时间优选为14~18h,进一步优选为15~17h。在本发明中,优选对步骤(3)中反应的产物体系进行过滤,所述过滤得到的棕色固体即为2-(2-乙氧基-2-氧乙基)-1-甲基-9h-吡啶并[3,4-b]吲哚-2-溴化铵。本发明还提供了一种由2-(2-乙氧基-2-氧乙基)-1-甲基-9h-吡啶并[3,4-b]吲哚-2-溴化铵制备得到的化合物,具有式2所示结构,本发明还提供了式2所述化合物的制备方法,反应步骤如下:将2-(2-乙氧基-2-氧乙基)-1-甲基-9h-吡啶并[3,4-b]吲哚-2-溴化铵、1,2-环己二酮、甲醇钠和二甲基甲酰胺混合后,反应温度为90~110℃,反应时间为14~18h,得到2,3,4,13-四氢-1h-吲哚并[2',3':3,4]吡啶并[1,2-b]异喹啉-6-溴化铵;所述反应的反应过程如下:在本发明中,所述步骤(a)反应温度为90~110℃,优选为95~105℃,更优选为98~102℃;所述反应时间为14~18h,优选为15~17h,更优选为15.5~16.5h。在本发明中,所述步骤(a)中2-(2-乙氧基-2-氧乙基)-1-甲基-9h-吡啶并[3,4-b]吲哚-2-溴化铵、1,2-环己二酮、甲醇钠和二甲基甲酰胺的用量比优选为1g:(0.3~0.5)g:(0.5~0.7)g:(13~15)ml,进一步优选为1g:(0.35~0.45)g:(0.55~0.65)g:(13.5~14.5)ml,更优选为1g:(0.38~0.42)g:(0.58~0.62)g:(13.8~14.2)ml。在本发明中,所述步骤(a)中甲醇钠优选在惰性气体下添加,所述惰性气体优选为氮气;所述甲醇钠加入时的温度优选为20~30℃,进一步优选为22~28℃,更优选为24~26℃。在本发明中,当所述步骤(a)的反应到达反应时间后,添加终止剂终止反应。在本发明中,所述步骤(a)中终止剂优选为乙酸,所述甲醇钠与终止剂的质量比优选为1:(1~1.2),进一步优选为1:(1.05~1.15)。在本发明中,优选对步骤(a)中终止后得到的产物体系进行液相色谱分离。在本发明中,所述步骤(a)中液相色谱分离的色谱柱优选为硅胶柱,所述硅胶柱的目数优选为230~400目,进一步优选为260~370目,更优选为300~330目;所述液相色谱分离的洗脱剂优选为含甲醇的二氯甲烷溶液,所述甲醇的百分比优选为0~10%,进一步优选为2~8%,更优选为4~6%。在本发明中,对步骤(a)中产物体系进行液相色谱分离后得到的黄绿固体即为2,3,4,13-四氢-1h-吲哚并[2',3':3,4]吡啶并[1,2-b]异喹啉-6-溴化铵。本发明还提供了一种由2-(2-乙氧基-2-氧乙基)-1-甲基-9h-吡啶并[3,4-b]吲哚-2-溴化铵制备得到的化合物,具有式3所示结构,本发明还提供了式3所述化合物的制备方法,反应步骤如下:将2-(2-乙氧基-2-氧乙基)-1-甲基-9h-吡啶并[3,4-b]吲哚-2-溴化铵、1,2-环己二酮、甲醇钠和二甲基甲酰胺混合后,反应温度为50~70℃,反应时间为0.5~1.5h,得到5-羧基-2,3,4,13-四氢-1h-吲哚并[2',3':3,4]吡啶并[1,2-b]异喹啉-6-溴化铵;所述反应的反应过程如下:在本发明中,所述步骤(b)中反应温度为50~70℃,优选为55~65℃,更优选为58~62℃;所述反应时间为0.5~1.5h,优选为0.8~1.2h,更优选为0.9~1.1h。在本发明中,所述步骤(b)中2-(2-乙氧基-2-氧乙基)-1-甲基-9h-吡啶并[3,4-b]吲哚-2-溴化铵、1,2-环己二酮、甲醇钠和二甲基甲酰胺的用量比优选为1g:(400~430)mg:(580~620)mg:(14~24)ml,进一步优选为1g:(410~420)mg:(590~610)mg:(16~22)ml,更优选为1g:(412~418)mg:(590~610)mg:(16~22)ml。在本发明中,所述步骤(b)中甲醇钠加入时的温度优选为20~30℃,进一步优选为22~28℃,更优选为24~26℃。在本发明中,当所述步骤(b)的反应到达反应时间后,添加终止剂终止反应。在本发明中,所述步骤(b)中终止剂优选为乙酸,所述甲醇钠与终止剂的质量比优选为1:(1~1.2),进一步优选为1:(1.05~1.15)。在本发明中,优选对步骤(b)中终止后得到的产物体系进行液相色谱分离。在本发明中,所述步骤(b)中液相色谱分离的色谱柱优选为硅胶柱,所述硅胶柱的目数优选为230~400目,进一步优选为260~370目,更优选为300~330目;所述液相色谱分离的洗脱剂优选为含甲醇的二氯甲烷溶液,所述甲醇的百分比优选为0~10%,进一步优选为2~8%,更优选为4~6%。在本发明中,对步骤(b)中产物体系进行液相色谱分离后得到的红色固体即为5-羧基-2,3,4,13-四氢-1h-吲哚并[2',3':3,4]吡啶并[1,2-b]异喹啉-6-溴化铵。本发明还提供了式1、式2或式3任意一种化合物在制备治疗癌症疼痛药物中的应用。下面结合实施例对本发明提供的技术方案进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。实施例1称取100gl-色氨酸和2423.73g乙醛,加入到500ml的乙酸中进行加成反应,温度为80℃,搅拌1h,得到的混合物减压浓缩,固体用500ml的水稀释,添加氨水调节ph为7,过滤收集固体,使用500ml的水和200ml的乙腈依次洗涤后得到的63g淡黄色固体即为3s-1-甲基-2,3,4,9-四氢-吡啶并[3,4-b]吲哚-3-羧酸。重复此步骤,制备更多的3s-1-甲基-2,3,4,9-四氢-吡啶并[3,4-b]吲哚-3-羧酸待用。3s-1-甲基-2,3,4,9-四氢-吡啶并[3,4-b]吲哚-3-羧酸的核磁共振氢谱结果如下:(400mhz,cd3od)δ13.48(s,1h),8.92–8.70(m,3h),8.52(d,j=8.0hz,1h),7.87(d,j=8.2hz,1h),7.75(ddd,j=8.2,7.0,1.2hz,1h),7.49(ddd,j=8.0,7.0,1.0hz,1h),4.24(s,3h),3.21(s,2h),2.92(s,2h),1.92(d,j=2.8hz,4h)。称取68g3s-1-甲基-2,3,4,9-四氢-吡啶并[3,4-b]吲哚-3-羧酸、149.33gn,n-二乙基乙胺、340ml二甲基甲酰胺和235.97gn-氯代丁二酰亚胺在0℃下混合均匀,所得混合物在25℃下搅拌30min,到达反应时间后添加1000ml的水淬灭反应,使用3×1000ml的乙酸乙酯萃取,得到的混合相使用3×1000ml的饱和碳酸钠溶液和800ml的盐水依次洗涤,洗涤得到的产物使用硫酸钠干燥,将上述体系减压过滤浓缩后得到的固体,使用500ml乙醚洗涤,得到的33.3g黄色固体即为1-甲基-9h-吡啶并[3,4-b]吲哚。重复此步骤,制备更多的1-甲基-9h-吡啶并[3,4-b]吲哚待用。1-甲基-9h-吡啶并[3,4-b]吲哚的核磁共振氢谱结果如下:(400mhz,cd3od)1hnmr(300mhz,dmso-d6)δ11.59(s,1h),8.21(dd,j=6.6,4.5hz,2h),7.93(d,j=5.4hz,1h),7.61(d,j=8.0hz,1h),7.54(ddd,j=8.2,6.8,1.2hz,1h),7.23(ddd,j=8.0,6.8,1.2hz,1h),2.78(s,3h)。取141g1-甲基-9h-吡啶并[3,4-b]吲哚和193.83g溴乙酸乙酯加入到1400ml丙酮中,在60℃下搅拌16h,过滤收集得到的205g棕色固体为2-(2-乙氧基-2-氧乙基)-1-甲基-9h-吡啶并[3,4-b]吲哚-2-溴化铵。2-(2-乙氧基-2-氧乙基)-1-甲基-9h-吡啶并[3,4-b]吲哚-2-溴化铵的核磁共振氢谱结果如下:(400mhz,cd3od)δ13.08(s,1h),8.72(d,j=6.4hz,1h),8.63(d,j=6.6hz,1h),8.47(d,j=8.2hz,1h),7.84–7.76(m,2h),7.46(ddd,j=8.0,4.6,3.3hz,1h),5.82(s,2h),4.26(q,j=7.2hz,2h),3.05(s,3h),1.25(t,j=7.2hz,3h)。取90g2-(2-乙氧基-2-氧乙基)-1-甲基-9h-吡啶并[3,4-b]吲哚-2-溴和37.55g1,2-环己二酮混合溶于预先加入54.31g甲醇钠的1289.00ml二甲基甲酰胺溶液中,在氮气氛围中置于25℃溶解;得到的混合溶液再置于100℃反应16h,到达反应时间后添加60g乙酸停止反应。将产物体系通过300目的硅胶快速色谱纯化,使用5%甲醇的二氯甲烷溶液作为洗脱剂,得到的26.3g黄色固体即为2,3,4,13-四氢-1h-吲哚并[2',3':3,4]吡啶并[1,2-b]异喹啉-6-溴化铵。2,3,4,13-四氢-1h-吲哚并[2',3':3,4]吡啶并[1,2-b]异喹啉-6-溴化铵的核磁共振氢谱结果如下:(400mhz,cd3od)δ13.48(s,1h),8.92–8.70(m,3h),8.52(d,j=8.0hz,1h),7.87(d,j=8.2hz,1h),7.75(ddd,j=8.2,7.0,1.2hz,1h),7.49(ddd,j=8.0,7.0,1.0hz,1h),4.24(s,3h),3.21(s,2h),2.92(s,2h),1.92(d,j=2.8hz,4h)。取1.05g2-(2-乙氧基-2-氧乙基)-1-甲基-9h-吡啶并[3,4-b]吲哚-2-溴和436.80mg1,2-环己二酮混合溶于预先加入631.8mg甲醇钠的15.00ml二甲基甲酰胺溶液中,置于25℃溶解;得到的混合溶液再置于60℃反应1h,到达反应时间后添加702mg乙酸停止反应。将产物体系通过300目的硅胶快速色谱纯化,使用5%甲醇的二氯甲烷溶液作为洗脱剂,得到的64.6mg红色固体即为5-羧基-2,3,4,13-四氢-1h-吲哚并[2',3':3,4]吡啶并[1,2-b]异喹啉-6-溴化铵。5-羧基-2,3,4,13-四氢-1h-吲哚并[2',3':3,4]吡啶并[1,2-b]异喹啉-6-溴化铵的核磁共振氢谱结果如下:(400mhz,cd3od)δ13.34(s,1h),8.62–8.53(m,3h),8.33(d,j=7.8hz,1h),7.79(d,j=8.0hz,1h),7.63(t,j=7.6hz,1h),7.40(dd,j=9.6,5.6hz,1h),3.13(s,2h),2.91(s,2h),1.85(dd,j=6.8,3.2hz,4h)。实施例2称取120gl-色氨酸和3060g乙醛,加入到636ml的乙酸中进行加成反应,温度为85℃,搅拌1.2h,得到的混合物减压浓缩,固体用624ml的水稀释,添加氨水调节ph为7,过滤收集固体,使用624ml的水和264ml的乙腈依次洗涤后得到的78g淡黄色固体即为3s-1-甲基-2,3,4,9-四氢-吡啶并[3,4-b]吲哚-3-羧酸。重复此步骤,制备更多的3s-1-甲基-2,3,4,9-四氢-吡啶并[3,4-b]吲哚-3-羧酸待用。称取70g3s-1-甲基-2,3,4,9-四氢-吡啶并[3,4-b]吲哚-3-羧酸、153.3gn,n-二乙基乙胺、357ml二甲基甲酰胺和256.2gn-氯代丁二酰亚胺在0℃下混合均匀,所得混合物在26℃下搅拌35min,到达反应时间后添加1054ml的水淬灭反应,使用3×1050ml的乙酸乙酯萃取,得到的混合相使用3×1050ml的饱和碳酸钠溶液和900ml的盐水依次洗涤,洗涤得到的产物使用硫酸钠干燥,将上述体系减压过滤浓缩后得到的固体,使用525ml乙醚洗涤,得到的36g黄色固体即为1-甲基-9h-吡啶并[3,4-b]吲哚。重复此步骤,制备更多的1-甲基-9h-吡啶并[3,4-b]吲哚待用。取120g1-甲基-9h-吡啶并[3,4-b]吲哚和168g溴乙酸乙酯加入到1300ml丙酮中,在65℃下搅拌15h,过滤收集得到的184g棕色固体为2-(2-乙氧基-2-氧乙基)-1-甲基-9h-吡啶并[3,4-b]吲哚-2-溴化铵。取50g2-(2-乙氧基-2-氧乙基)-1-甲基-9h-吡啶并[3,4-b]吲哚-2-溴和23g1,2-环己二酮混合溶于预先加入33g甲醇钠的700.00ml二甲基甲酰胺溶液中,在氮气氛围中置于24℃溶解;得到的混合溶液再置于105℃反应15h,到达反应时间后添加36.3g乙酸停止反应。将产物体系通过300目的硅胶快速色谱纯化,使用4%甲醇的二氯甲烷溶液作为洗脱剂,得到的13.7g黄色固体即为2,3,4,13-四氢-1h-吲哚并[2',3':3,4]吡啶并[1,2-b]异喹啉-6-溴化铵。取12g2-(2-乙氧基-2-氧乙基)-1-甲基-9h-吡啶并[3,4-b]吲哚-2-溴和4.98g1,2-环己二酮混合溶于预先加入7.14g甲醇钠的204.00ml二甲基甲酰胺溶液中,置于24℃溶解;得到的混合溶液再置于55℃反应0.8h,到达反应时间后添加8.2g乙酸停止反应。将产物体系通过300目的硅胶快速色谱纯化,使用4%甲醇的二氯甲烷溶液作为洗脱剂,得到的738.28mg红色固体即为5-羧基-2,3,4,13-四氢-1h-吲哚并[2',3':3,4]吡啶并[1,2-b]异喹啉-6-溴化铵。实施例3称取80gl-色氨酸和2080g乙醛,加入到320ml的乙酸中进行加成反应,温度为75℃,搅拌1.2h,得到的混合物减压浓缩,固体用320ml的水稀释,添加氨水调节ph为7,过滤收集固体,使用340ml的水和200ml的乙腈依次洗涤后得到的48g淡黄色固体即为3s-1-甲基-2,3,4,9-四氢-吡啶并[3,4-b]吲哚-3-羧酸。重复此步骤,制备更多的3s-1-甲基-2,3,4,9-四氢-吡啶并[3,4-b]吲哚-3-羧酸待用。称取42g3s-1-甲基-2,3,4,9-四氢-吡啶并[3,4-b]吲哚-3-羧酸、98.82gn,n-二乙基乙胺、197.6ml二甲基甲酰胺和172.9gn-氯代丁二酰亚胺在0℃下混合均匀,所得混合物在26℃下搅拌25min,到达反应时间后添加680ml的水淬灭反应,使用3×700ml的乙酸乙酯萃取,得到的混合相使用3×700ml的饱和碳酸钠溶液和600ml的盐水依次洗涤,洗涤得到的产物使用硫酸钠干燥,将上述体系减压过滤浓缩后得到的固体,使用300ml乙醚洗涤,得到的20.6g黄色固体即为1-甲基-9h-吡啶并[3,4-b]吲哚。重复此步骤,制备更多的1-甲基-9h-吡啶并[3,4-b]吲哚待用。取160g1-甲基-9h-吡啶并[3,4-b]吲哚和224g溴乙酸乙酯加入到1440ml丙酮中,在55℃下搅拌15h,过滤收集得到的232g棕色固体为2-(2-乙氧基-2-氧乙基)-1-甲基-9h-吡啶并[3,4-b]吲哚-2-溴化铵。取75g2-(2-乙氧基-2-氧乙基)-1-甲基-9h-吡啶并[3,4-b]吲哚-2-溴和30g1,2-环己二酮混合溶于预先加入48.75g甲醇钠的1050.00ml二甲基甲酰胺溶液中,在氮气氛围中置于26℃溶解;得到的混合溶液再置于108℃反应18h,到达反应时间后添加55.23g乙酸停止反应。将产物体系通过300目的硅胶快速色谱纯化,使用6%甲醇的二氯甲烷溶液作为洗脱剂,得到的22.3g黄色固体即为2,3,4,13-四氢-1h-吲哚并[2',3':3,4]吡啶并[1,2-b]异喹啉-6-溴化铵。取3.3g2-(2-乙氧基-2-氧乙基)-1-甲基-9h-吡啶并[3,4-b]吲哚-2-溴和1.40g1,2-环己二酮混合溶于预先加入2029.5mg甲醇钠的76.00ml二甲基甲酰胺溶液中,置于26℃溶解;得到的混合溶液再置于65℃反应1.3h,到达反应时间后添加702mg乙酸停止反应。将产物体系通过300目的硅胶快速色谱纯化,使用5%甲醇的二氯甲烷溶液作为洗脱剂,得到的64.6mg红色固体即为5-羧基-2,3,4,13-四氢-1h-吲哚并[2',3':3,4]吡啶并[1,2-b]异喹啉-6-溴化铵。应用实施例本应用实施例采用实施例1中制备得到的2,3,4,13-四氢-1h-吲哚并[2',3':3,4]吡啶并[1,2-b]异喹啉-6-溴化铵,为便于区分,记为生物碱a。实验一、生物碱a对化学与物理因素引起的小鼠镇痛实验研究。1.动物分组及给药5周龄icr小鼠,雄性雌性各50只,体重18-24g,实验前先适应性喂养7天。称取生物碱a适量用超纯水溶解,配制成混悬液。然后按照剂量设定,各给药组灌胃给药;模型组灌胃给予等体积生理盐水。小鼠给药体积为0.1ml/10g。2.实验方法及数据处理2.1生物碱a对化学因素致痛的影响采用小鼠腹腔注射化学药物的方法,引起腹腔深部较持久的疼痛刺激,使小鼠产生“扭体”反应,通过给药后测量痛阈值的高低而反映药物的镇痛作用。取雄性icr小鼠50只,按体重分层随机分成5组,每组10只,对照组给予生理盐水30ml/kg,生物碱a分小、中、大三个剂量给药,分别为3mg/kg、6mg/kg、12mg/kg。各组灌胃给药30min后,腹腔注射0.6%醋酸生理盐水溶液0.1ml/10g,记录自注射醋酸致痛后每只小鼠在15min内出现的扭体反应次数,扭体反应观测指标为腹部收缩内凹、躯干与后腿伸张、臀部抬高、蠕行,计算各给药组的扭体抑制率。扭体抑制率=[(对照组扭体次数-药物组扭体次数)/对照组扭体次数]×100%实验数据以x±s表示,采用spss21.0软件进行方差分析。2.2生物碱a对物理因素致痛的影响小鼠的足部接触热板,使其受热刺激而产生疼痛反应,通过给药后测量不同时间内痛阈值的大小而反映药物的镇痛作用。取雌性icr小鼠50只,按体重分层随机分成5组,每组10只,对照组给予生理盐水30ml/kg,生物碱a分小中大剂量给药,分别为3mg/kg、6mg/kg、12mg/kg。将小鼠置于ysd-ix药理热板仪中,调节温度55℃,观察记录小鼠在不同时间内接触热板至舔后足的时间。3.实验结果表1各组药物对化学因素致痛引起的影响(x±s)组别剂量(mg/kg)扭体次数抑制率模型组38.6±3.9a—生物碱a1组321.2±8.9**45.1%生物碱a2组626.6±9.4**a31.1%生物碱a3组1215.8±7.5**59.1%表2各组药物对物理因素致痛引起的影响(x±s)备注:与模型组比较,**p<0.01;由表1所示的实验结果可知生物碱a有显著的镇痛效果,并呈现量效正相关,可明显减少化学刺激引起的扭体次数和延长小鼠舔足反应的潜伏期,具有较强的镇痛作用。实验二、生物碱a对人神经胶质瘤(u251)移植瘤的生长抑制实验研究实验动物野生型ab品系斑马鱼,以自然成对交配繁殖方式进行,共330尾,每实验组为30尾,年龄为3dpf,用于生物碱a最大检测浓度(mtc)测定。野生型ab品系斑马鱼,以自然成对交配繁殖方式进行,共150尾,每实验组为30尾,年龄为2dpf,用于生物碱a肿瘤生长抑制作用评价。饲养于28℃的养鱼用水中(水质:每1l反渗透水中加入200mg速溶海盐,电导率为480~510μs/cm;ph为6.9~7.2;硬度为53.7~71.6mg/lcaco3),实验动物使用许可证号为:syxk(浙)2012-0171。饲养管理符合国际aaalac认证的要求。实验药物生物碱a,纯度99.0%,棕色粉末,阴凉柜干燥保存,由福建中医药大学药学院3408实验室提供。临用前dmso配成20mg/ml母液,-20℃保存。仪器与试剂解剖显微镜(szx7,olympus,japan);ccd相机(verta1,上海土森视觉科技有限公司);显微注射仪(im300,narishige,japan);电动聚焦连续变倍荧光显微镜(az100,nikon,japan);精密电子天平(cp214,ohaus,america);6孔板(nestbiotech,shanghai,china)。浓度组别确定最大检测浓度(mtc)抗肿瘤作用评价浓度确定依据根据浓度摸索实验,生物碱a对斑马鱼肿瘤生长抑制作用的mtc为3.13μg/ml。故此,生物碱a的实验浓度设置为:0.35、1.04和3.12μg/ml。模型制作用荧光染料标记的人神经胶质瘤(u251)细胞,以显微注射的方式移植到斑马鱼卵黄囊内,每尾移植约200个细胞,建立斑马鱼肿瘤移植模型。实验方法1.mtc的测定随机选取330尾3dpf野生型ab品系斑马鱼于六孔板中,每孔(实验组)均处理30尾斑马鱼,分别水溶暴露给予生物碱a0.39、0.78、1.56、3.12、6.25、12.5、25、50、100和200μg/ml,同时设置正常对照组,每孔(实验组)容量为3ml。35℃培养箱培养至5dpf,统计斑马鱼的死亡情况,根据斑马鱼毒性反应和死亡情况确定生物碱a对斑马鱼的mtc。2.对u251移植瘤的生长抑制作用评价使用荧光染料标记的人神经胶质瘤(u251)细胞,以显微注射的方式移植到受精后2天(2dpf)野生型ab品系斑马鱼卵黄囊内,每尾移植约200个细胞,建立斑马鱼肿瘤移植模型;将注射神经胶质瘤细胞的斑马鱼放置35℃培养至3dpf。3dpf时,在显微镜下挑选出移植瘤一致性较好的斑马鱼,随机分配至6孔板中,每孔30尾,每孔养鱼水容量为3ml。水溶暴露给予生物碱a0.35、1.04和3.12μg/ml浓度,阳性对照药顺铂15μg/ml浓度,同时设置模型对照组。35℃培养箱处理至5dpf,每个实验组随机选择10尾斑马鱼在荧光显微镜下采集斑马鱼肿瘤细胞荧光强度(s),以荧光强度的统计分析结果评价生物碱a对斑马鱼u251移植瘤的生长抑制作用。统计学处理结果用mean±se表示,对斑马鱼肿瘤生长的抑制作用计算公式如下:统计学分析采用单因素方差分析和dunnett’st检验,p<0.05表明具有显著性差异,提供具有代表性的实验图谱。实验结果1.mtc生物碱a在0.39~3.12μg/ml浓度时,对斑马鱼未产生任何明显的毒副作用,也未引起斑马鱼死亡;在6.25μg/ml浓度时,诱发3尾死亡;在12.5μg/ml浓度时,诱发15尾死亡;在25~200μg/ml浓度时,诱发100%斑马鱼死亡。因此,确定生物碱a对斑马鱼肿瘤生长抑制作用的mtc为3.12μg/ml。详见表3。表3.生物碱a处理后“浓度-致死性”结果(n=30)2.对u251移植瘤的抑制作用结果显示:阳性对照药顺铂15μg/ml浓度斑马鱼u251移植瘤荧光强度为145631像素,与模型对照组(320746像素)比较p<0.001,肿瘤生长抑制作用为55%,表明顺铂可明显抑制u251移植瘤生长。生物碱a0.35、1.04和3.12μg/ml浓度组斑马鱼u251移植瘤荧光强度分别为325368、246081和133109像素,与模型对照组(320746像素)比较p>0.05&p<0.05&p<0.001,肿瘤生长抑制作用分别为-1%、23%和59%。表明生物碱a在1.04和3.12μg/ml浓度条件下具有抗神经胶质瘤(u251)作用。详见表4、图3、图4和图5。表4.生物碱a碱对斑马鱼u251移植瘤的抑制作用(n=10)与模型对照组比较,*p<0.05,***p<0.001实验结论在本实验浓度条件下,生物碱a对人神经胶质瘤(u251)细胞移植瘤的生长具有抑制作用。实验三、生物碱a对人鼻咽癌细胞(cne2luc)移植瘤的生长抑制实验研究1实验目的通过注射cne2luc(荧光素酶标记的低分化鼻咽癌)细胞入3-4天斑马鱼卵黄囊中建立斑马鱼荷瘤模型,给药干预后观察荧光强度的变化,进行生物碱a的体内抗鼻咽癌的药效评价。2实验方法2.1生物碱a对斑马鱼幼鱼存活率及致畸的影响取刚孵化3-4天的斑马鱼幼鱼120条,随机分组10组,分别为空白对照组,0.5%dmso组以及给药组(800、400、200、100、50、25、12.5、6.25μmol/l)每组12条,用胚胎水配制成相应的药物,使鱼生活在相应浓度的药液中,48h后观察其存活率及致畸情况。2.2生物碱a对荷瘤斑马鱼的影响取培养致对数生长期的cne2luc细胞,消化后用细胞荧光染料将细胞转染荧光标记,之后用无血清rpmi-1640培养基将浓度调整至1x107个/ml,按200个/鱼(2nl)的剂量将细胞悬液注射进3-4天的幼鱼中,注射细胞4h后选取荧光强度相近的鱼进行试验分5组,分组后给药前各条进行拍照,按编号放入96孔板内,每孔1条,分别给予各组0.5%dmso组和生物碱a给药组25μmol/l、12.5μmol/l、6.25μmol/l剂量组,每组8条,按2.1项下方法给药,48h后观察荷瘤斑马鱼携带的荧光标记强度,本鱼给药前后拍照时其曝光时间及拍照体位保持一致,将拍照图片使用imagej软件进行处理,与本鱼进行前后变化比较,判断药物的效用,结果以荧光总强度降低率表示。3实验结果3.1生物碱a对正常斑马鱼幼鱼的存活率的影响生物碱a对正常斑马鱼幼鱼的存活率结果见图6,由图可知,当给药剂量为50μmol/l时其存活率为50%,及生物碱a对正常斑马鱼幼鱼48h的ld50为50μmol/l,后续实验中,选取25μmol/l、12.5μmol/l、6.25μmol/l进行。3.2生物碱a对荷瘤斑马鱼的影响从结果可知,生物碱a可一定程度上降低荷瘤斑马鱼的荧光强度,三个剂量组较空白对照组有显著性差异,但药物毒性较大,25μmol/l浓度下对荷瘤斑马鱼有较大的毒性,且每组样本量较少,具体实验结果如表5及图7表5各组斑马鱼给药前后自身荧光强度降低率(%)4.实验结论在本实验条件下,生物碱a对人鼻咽癌细胞(cne2luc)移植瘤的生长具有抑制作用。采用本发明实施例2和实施例3制备得到的最终化合物也能达到和实施例1相似的效果。由以上实施例可知,本发明提供了一种可有效对抗癌症及疼痛治疗的化合物,能明显的降低因物理或化学造成的疼痛,并很好的抑制癌细胞的生长。以上所述仅是本发明的优选实施方式,应当指出,对于本
技术领域:
的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1