一种诊断脊髓性肌萎缩症的CRISPR-Cas系统及其应用的制作方法
本发明涉及一种crispr-cas系统,特别是一种诊断脊髓性肌萎缩症的crispr-cas系统及其应用。
背景技术:
sma是一类由于脊髓前角运动神经元变性导致的对称性肌无力和肌萎缩的神经肌肉疾病,是一种婴儿期最常见的常染色体隐性遗传性疾病之一,主要表现为肢体近端肌无力,随病情加重,躯体运动功能下降或丧失,出现吞咽和自主呼吸困难,最终因呼吸肌麻痹而死亡。sma在人群中的发病率约为1/6000-1/10000[1]。通常根据疾病严重程度及发病年龄主要将sma分为三个亚型,其中sma-i型约占50%,患者在出生时或在出生6个月内发病,不能独自坐或走,通常会在两年内因呼吸肌麻痹而死亡[2]。sma属于严重致死、致残的遗传性疾病,给患者家庭及社会都带来了巨大的负担,已经成了亟待解决的公共卫生问题。
sma的致病基因是编码运动神经元存活蛋白(survivalmotorneuron,smn)的smn1基因,人的smn基因定位于5q11.2-5q13.3[3],且有两个高度同源的拷贝,靠近端粒端的称为smn1,而靠近着丝粒的称为smn2,二者在编码序列区只相差一个碱基,在smn1基因7号外显子第6位碱基为c,而smn2该位置为t,smn2基因产生了选择性剪接,翻译出的蛋白质约有90%是截短型蛋白,不能正常发挥功能[4]。有研究显示smn1基因7号外显子纯合缺失出现在95%以上的sma患者中,是sma的热点致病突变[5,6]。目前已经有nusinersen和zolgensma获批用于治疗sma。对sma患者准确的遗传诊断,有利于患儿的早发现、早干预、早治疗,而且还可以在植入前诊断与产前诊断中阻止患儿的出生,因此,开发一种快速准确、简便廉价且易于普及的检测方法应用于sma的遗传诊断,从而有效的防治sma,具有重大的社会价值与现实意义。
针对smn1基因的检测技术是开展sma诊断的前提,目前已经有一些技术用于sma诊断。聚合酶链反应-单链构象多态性分析(polymerasechainreaction-singlestrandconformationpolymorphism,pcr-sscp)和pcr-限制性片段长度多态性分析(pcr-restrictionfragmentlengthpolymorphism,pcr-rflp)操作比较繁琐,且可能会因为酶切不完全导致假阳性[3,7];pcr-变性高效液相色谱(pcr-denaturehighperformanceliquidchromatography)高度依赖于仪器设备[8];定量pcr技术对样本dna质量要求较高,且检测成本较高[9];多重连接依赖探针扩增技术(multiplexligation-dependentprobeamplification,mlpa)操作繁琐,对仪器设备和操作人员要求高,且检测耗时较长[10];高分辨熔解曲线和液滴数字pcr均对仪器设备要求较高。目前这些检测方法在原理、适用范围、检测性能和检测成本等方面各有差异,但是操作繁琐,且对仪器设备和操作人员要求较高,不便于推广普及。
普遍存在于细菌和古细菌中的成簇规律间隔短回文重复序列(clusteredregularlyinterspacedshortpalindromerepeats,crispr)和crispr相关核酸酶(crisprassociatedproteins,cas)是细菌抵抗外源感染的防御体系[11],cas蛋白能在crisprrna(crrna)的引导下特异识别和切割靶dna。2016年fengzhang等发现cas13a(又名c2c2)在crrna引导下识别切割靶rna序列后能被激活,活化后的cas13a表现出不加区别的切割活性(附带切割活性),既能特异地切割靶rna分子,又能非特异地切割其他非靶rna分子[12]。2017年fengzhang等报道基于cas13a这一特性的rna分子检测技术,cas13a-crrna复合物特异识别切割扩增后的靶rna分子,激活cas13a,活化后的cas13a非特异地切割rna荧光探针,通过检测荧光信号达到检测靶rna的目的[13]。
cas蛋白家族的另一成员cas12a(又名cpf1)能在crrna引导下识别切割靶双链dna(doublestrandeddna,dsdna)并被激活[14,15]。2018年chen和li分别报道基于cas12a的dna分子检测技术[14,16]。目前cn110551812a利用crispr/cas12a靶向核酸检测技术诊断sma,主要流程是通过pcr扩增包含smn1基因7号外显子区序列,琼脂糖胶电泳之后切胶回收,然后利用酶标仪每1分钟检测1次荧光。但此方法中crrna同时会识别smn1和smn2基因7号外显子,当sma患者缺失smn1基因,含有smn2基因时,此方法不会将其判定为sma患者,但是几乎所有的sma患者至少有1个拷贝的smn2基因,故其准确性难以保证。同时该方法操作繁琐、费时、费力不利于进行大量样本的同时检测。
cas14a1是新近发现的cas蛋白家族的又一成员,无需pam序列即可在crrna引导下特异识别和切割靶ssdna并被激活,激活后的cas14a1蛋白具有非特异性切割ssdna的附带切割活性。2018年doudna等报道基于cas14a1的dna分子检测技术,通过采用硫代磷酸酯修饰的引物扩增dna后,加入t7核酸外切酶处理获得ssdna,靶ssdna被cas14a1-crrna复合物特异识别切割,从而激活cas14a1,活化后cas14a1的附带切割效应切割ssdna荧光探针,通过检测荧光信号从而达到检测靶dna的目的[17]。但是通过两步才能获得单链的ssdna,步骤繁琐,而频繁的开盖会增加污染发生风险。且采用的引物需要特殊的修饰,引物价格昂贵。cas14a1灵敏度高,检测成本低,具有更高的特异性,无需引入错配即可高效区分单个碱基的差异;且无pam序列限制,可特异性检测任意与crrna互补配对的ssdna。这有效地克服了现阶段sma检测技术存在的不足,可实现对smn1基因7号外显子检测。目前国际上尚无crispr/cas14a1分子检测技术应用于遗传病诊断的报道。
参考文献:
[1]j.pearn,classificationofspinalmuscularatrophies[j].lancet,1980,1:919-922.
[2]e.mercuri,e.bertini,s.t.iannaccone,childhoodspinalmuscularatrophy:controversiesandchallenges[j].thelancet.neurology,2012,11:443-452.
[3]s.lefebvre,l.burglen,s.reboullet,etal.,identificationandcharacterizationofaspinalmuscularatrophy-determininggene[j].cell,1995,80:155-165.
[4]b.wirth,anupdateofthemutationspectrumofthesurvivalmotorneurongene(smn1)inautosomalrecessivespinalmuscularatrophy(sma)[j].humanmutation,2000,15:228-237.
[5]a.h.burghes,c.e.beattie,spinalmuscularatrophy:whydolowlevelsofsurvivalmotorneuronproteinmakemotorneuronssick?[j].naturereviews.neuroscience,2009,10:597-609.
[6]l.alias,s.bernal,p.fuentes-prior,etal.,mutationupdateofspinalmuscularatrophyinspain:molecularcharacterizationof745unrelatedpatientsandidentificationoffournovelmutationsinthesmn1gene[j].humangenetics,2009,125:29-39.
[7]g.vandersteege,p.m.grootscholten,p.vandervlies,etal.,pcr-baseddnatesttoconfirmclinicaldiagnosisofautosomalrecessivespinalmuscularatrophy[j].lancet,1995,345:985-986.
[8]r.sutomo,t.akutsu,y.takeshima,etal.,rapidsmn1deletiontestusingdhplctoscreenpatientswithspinalmuscularatrophy[j].americanjournalofmedicalgenetics,2002,113:225-226.
[9]m.feldkotter,v.schwarzer,r.wirth,etal.,quantitativeanalysesofsmn1andsmn2basedonreal-timelightcyclerpcr:fastandhighlyreliablecarriertestingandpredictionofseverityofspinalmuscularatrophy[j].amjhumgenet,2002,70:358-368.
[10]c.h.huang,y.y.chang,c.h.chen,etal.,copynumberanalysisofsurvivalmotorneurongenesbymultiplexligation-dependentprobeamplification[j].geneticsinmedicine:officialjournaloftheamericancollegeofmedicalgenetics,2007,9:241-248.
[11]y.ishino,h.shinagawa,k.makino,etal.,nucleotidesequenceoftheiapgene,responsibleforalkalinephosphataseisozymeconversioninescherichiacoli,andidentificationofthegeneproduct[j].journalofbacteriology,1987,169:5429-5433.
[12]a.east-seletsky,m.r.o'connell,s.c.knight,etal.,twodistinctrnaseactivitiesofcrispr-c2c2enableguide-rnaprocessingandrnadetection[j].nature,2016,538:270-273.
[13]j.s.gootenberg,o.o.abudayyeh,j.w.lee,etal.,nucleicaciddetectionwithcrispr-cas13a/c2c2[j].science,2017,356:438-442.
[14]j.s.chen,e.ma,l.b.harrington,etal.,crispr-cas12atargetbindingunleashesindiscriminatesingle-strandeddnaseactivity[j].science,2018,360:436-439.
[15]s.y.li,q.x.cheng,j.k.liu,etal.,crispr-cas12ahasbothcis-andtrans-cleavageactivitiesonsingle-strandeddna[j].cellres,2018,28:491-493.
[16]s.y.li,q.x.cheng,j.m.wang,etal.,crispr-cas12a-assistednucleicaciddetection[j].celldiscov,2018,4:20.
[17]l.b.harrington,d.burstein,j.s.chen,etal.,programmeddnadestructionbyminiaturecrispr-cas14enzymes[j].science,2018,362:839-842.
技术实现要素:
本发明所要解决的技术问题是,本发明为了克服上诉技术方法的缺点,通过联合不对称pcr技术与crispr/cas14a1靶向核酸检测方法,建立了基于荧光变化值来实现低成本、高检测性能的诊断脊髓性肌萎缩症的crispr-cas系统。
为解决上述技术问题,本发明所采用的技术方案是:
一种诊断脊髓性肌萎缩症的crispr-cas系统,包括cas14a1。
优选的,所述crispr-cas系统包括cas14a1、crrna、ssdna和fq-probe。优选的,所述crrna序列如seqidno.1所示;所述gdna扩增引物包括如seqidno.2所示的上游引物和seqidno.3所示的下游引物;所述fq-probe序列如seqidno.4所示,且fq-probe的5’端修饰有荧光基团,3’端修饰有淬灭基团。
seqidno.1:
5'-taatacgactcactataggttcactgataaagtggagaaccgcttcaccaaaagctgtcccttaggggattagaacttgagtgaaggtgggctgcttgcatcagcctaatgtcgagaagtgctttcttcggaaagtaaccctcgaaacaaattcatttgaaagaatgaaggaatgcaacattttgtctgaaaccctgtaa-3';
seqidno.2:5'-aaataagtaaaatgtcttgtgaaac-3';
seqidno.3:5'-gaatgtgagcaccttccttctt-3';
seqidno.4:5'fam-ttttttttttttttttttttt-3'bhq-x。
所述fq-probe序列与现有技术中公开的序列不同。
优选的,所述crrna制备方法包括以下步骤:(1)设计并合成序列如seqidno.1所示的crrna;(2)用转录试剂盒对步骤(1)获得的crrna进行转录,即得所述crrna。
优选的,所述ssdna制备方法包括以下步骤:提取待检测样本dna,然后利用如seqidno.2所示的上游引物和seqidno.3所示的下游引物进行不对称pcr扩增,不对称pcr扩增产物即为所述ssdna。
本发明还提供了所述的诊断脊髓性肌萎缩症的crispr-cas系统在制备脊髓性肌萎缩症诊断试剂盒中的用途。
本发明提供的crispr-cas系统可用于特异性检测smn1基因的突变情况,且与现有技术不同之处在于,本发明还能区别开smn2基因干扰,从而判断被检测者是否患脊髓性肌萎缩症,因此,可用于制备脊髓性肌萎缩症诊断试剂盒。
本发明还提供了一种脊髓性肌萎缩症诊断试剂盒,包含权利要求1~5任一项所述的诊断脊髓性肌萎缩症的crispr-cas系统。
本发明还通过比较不同buffer产生的荧光增加值得到最佳使用buffer为neb的cutsmartbuffer(配方为:50mmkac,20mmtris-ac,10mmmg(ac)2,100μg/mlbsa(ph7.9@25℃))。
优选的,所述试剂盒包括:cas14a1100nm、crrna125nm、ssdna50ng、fq-probe0.6μl。
本发明还提供了所述的脊髓性肌萎缩症诊断试剂盒在制备脊髓性肌萎缩症诊断试剂中的用途。
下面对本发明做进一步的解释:
本发明利用crispr/cas14a1分子检测技术原理并优化ssdna的获取方式,建立针对smn1基因7号外显子的基因检测技术。设计识别smn1基因7号外显子的crrna与不对称pcr扩增引物,当模板dna中存在smn1基因7号外显子,经不对称pcr扩增后,cas14a1可在crrna引导下特异性的识别并切割ssdna产物中的smn1基因7号外显子而被激活,活化后的cas14a1通过附带切割效应,非特异性切割荧光探针,产生荧光信号;而当模板dna中不存在smn1基因7号外显子,仅存在smn2基因7号外显子时,由于smn2基因7号外显子与crrna存在碱基错配,不对称pcr扩增后的ssdna不能有效激活cas14a1,荧光探针未被切割而无荧光信号产生,通过读取荧光值便可判断是否为sma患者。
与现有技术相比,本发明所具有的有益效果为:
本发明基于crispr/cas14a1靶向核酸检测技术诊断sma,有以下几个优点:
(1)crrna不受pam序列的限制,可以自由设计;
(2)通过不对称pcr扩增的目的片段产物,可以直接用于检测,不需要经过胶回收,大大节约操作时间与成本;
(3)不需要持续性检测荧光,只需检测并记录初始荧光值以及1小时的荧光值即可;
(4)本发明中crrna可以特异性识别smn1基因7号外显子,而现有技术“一种诊断脊髓性肌萎缩症的crispr/cas系统及其应用”中crrna同时识别smn1和smn2基因7号外显子,故其方法难以有效的检测出含有smn2基因的sma患者,且几乎所有的sma患者至少有1个拷贝的smn2基因,因此本发明可使检测特异性和准确性大大提高。
至今尚没有联合不对称pcr技术与crispr/cas14a1检测技术的研究报道,也无crispr/cas14a1应用于sma患者基因检测的报道。本发明构建的基于crispr/cas14a1与不对称pcr结合的方法可进行sma患者基因检测,且具有操作简便、特异性高、低成本的特点。目前基于cas14a1的核酸检测技术都需使用硫代磷酸酯修饰的引物来扩增靶位点序列,然后用t7核酸外切酶处理获得ssdna,引物修饰昂贵,且增加了操作步骤,增大了污染风险,而本发明中我们创新性的将不对称pcr技术与cas14a1相结合,大大减少操作步骤与时间及检测成本,且在实施例中我们比较了联合t7核酸外切酶获得ssdna与不对称pcr扩增获得ssdna的crispr/cas14a1切割检测效果,不对称pcr与cas14a1相结合可以更加显著区分sma患者与非患者,在一定程度上提高了检测特异性和准确性。
附图说明
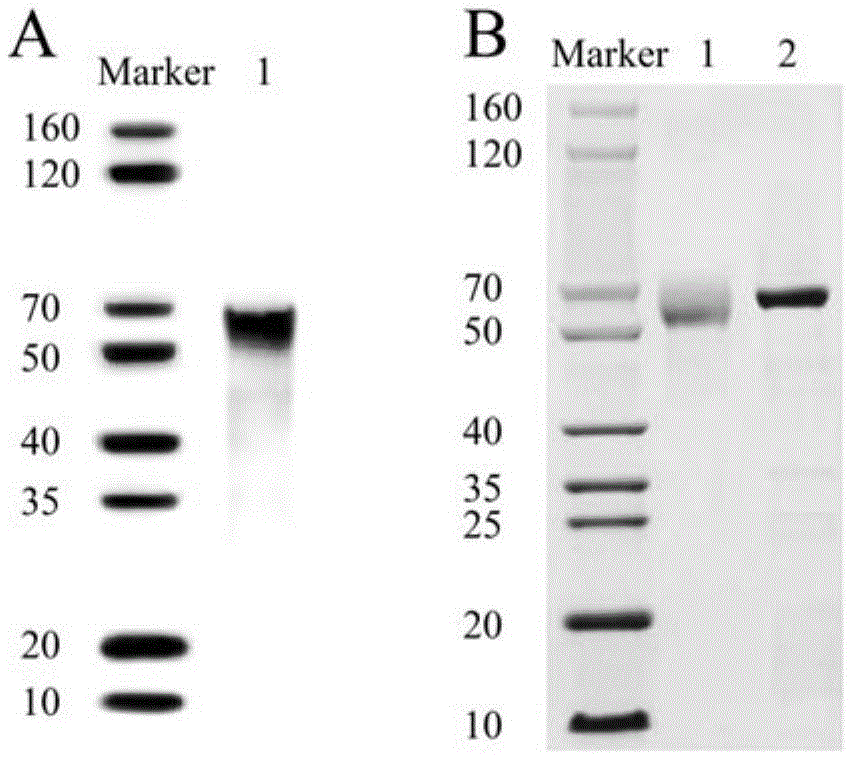
图1为本发明实施例1纯化后cas14a1蛋白的鉴定;其中图a为westernblot鉴定cas14a1蛋白;图b为sds-page胶电泳来评估cas14a1蛋白的纯度,泳道1为bsa作为对照;
图2为不对称pcr扩增的热循环条件图;
图3为crispr/cas14a1检测体系针对sma检测;blank为空白对照,ns,nosignificance,***,p<0.001;
图4为crispr/cas14a1检测体系对比不同长度的fq-probe的检测;
图5为先用硫代磷酸化修饰的pcr扩增后经t7核酸外切酶处理之后再进行crispr/cas14a1切割的检测结果,*,p<0.05;
图6为crispr/cas14a1检测sma检测下限;比较sma患者与非患者的荧光增加值,studenttest进行统计分析,*,p<0.05,ns,nosignificance;
图7为crispr/cas14a1检测临床样本结果;one-wayanova进行统计分析,***,p<0.0001。
具体实施方式
以下将结合实施例来详细说明本发明。需要说明的是,在不冲突的情况下,本发明中的实施例及实施例中的特征可以相互组合。
实施例1
cas14a1蛋白表达与纯化
构建cas14a1表达载体并利用大肠杆菌表达cas14a1蛋白,通过亲和层析进行纯化,免疫印迹检测cas14a1蛋白。
1)cas14a1蛋白表达
①将构建好的含有cas14a1基因的质粒转化到bl21(de3)感受态细胞中,均匀涂布到含50μg/ml的硫酸卡那霉素的lb平板上,倒置于37℃培养箱过夜培养。从转化的平板中挑选单克隆菌落,接种到4ml含50μg/ml硫酸卡那霉素的液体lb培养基中培养;
②待培养至od600为0.5-0.8,向lb培养液中加入终浓度为0.2mm的iptg,之后分别置于15℃、37℃条件下16小时诱导表达;
③取诱导后的培养液12000rpm离心5min,去除上清液,加入pbs重悬沉淀,最后加入sds-page上样缓冲液于100℃下加热样品10min,离心后取上清电泳。160v恒压电泳至溴酚蓝带迁移至离凝胶底部1cm时,取出凝胶利用蛋白凝胶快速处理系统染脱色,根据结果分析约63.8kd大小处蛋白条带来选取合适的诱导条件;
④确认蛋白可以诱导表达之后,进行蛋白扩大培养表达,培养1.6l的表达菌,生长至od600为0.8时,加入终浓度为0.2mm的iptg,15℃诱导表达16小时后收集菌体,用于后续蛋白纯化。
2)cas14a1蛋白纯化与鉴定
收集的全菌采用50mmtris(ph8.0),300mmnacl,20mmimidazole含1%tritonx-100,1mmdtt,1mmpmsf超声裂解,同时以50mmtris(ph8.0),300mmnacl,20mmimidazole缓冲液平衡ni-ida亲和层析柱,之后用不同浓度imidazole的平衡缓冲液洗脱目标蛋白,并收集每个洗脱组分进行sds-page分析检测,显示500mmimidazole平衡缓冲液洗脱的目标蛋白浓度相对较高,收集之后,将其透析到20mmhepes(ph7.5),1250mmnacl,10%glycerol中,透析结束后用0.22μm滤器过滤,并分装冻于-80℃。
利用anti-his抗体进行westernblot检测,结果显示成功表达cas14a1蛋白(图1a),同时r250染色的sds-page电泳表明所获得的cas14a1蛋白纯度大于90%(图1b),为后续建立基于cas14a1靶向核酸检测技术体系提供可靠蛋白来源。
实施例2
对已经明确基因型的dna进行检测,进行方法的验证
以正常人gdna为模板,pcr扩增之后进行crispr/cas14a1切割检测,通过比较不同buffer产生的荧光增加值得到最佳使用buffer为neb的cutsmartbuffer(配方为:50mmkac,20mmtris-ac,10mmmg(ac)2,100μg/mlbsa(ph7.9@25℃))。后续检测过程中我们均选cutsmartbuffer用作缓冲液。
1.crrna转录
(1)设计并合成如上表所示的crrna对应dna序列,并在体外利用hiscribetmt7highyieldrnasynthesiskit(newenglandbiolabs,e2040s)转录试剂盒进行转录获得crrna。
转录体系如下:
37℃孵育16小时。
(2)通过dnasei去除转录体系中可能残留的dna
37℃孵育30分钟。
(3)纯化crrna(利用rna纯化试剂盒进行)
①第(2)步体系中加入500μl无水乙醇,混匀后加入至纯化柱中,以9000g离心15秒;
②弃去收集管中液体,加入700μlrwt,以9000g离心15秒;
③弃去收集管中液体,加入500μlrpe,以9000g离心15秒;
④弃去收集管中液体,加入500μlrpe,以9000g离心2分钟;
⑤弃去收集管中液体,以9000g离心2分钟,干燥滤膜;
⑥将柱子装入新的1.5ml收集管,加入30-50μlrnase-freewater,以9000g离心1分钟,洗脱crrna,测浓度,分装后保存只-80℃冰箱待用。
2.不对称pcr扩增目的片段
利用seqidno.2所示的上游引物和seqidno.3所示的下游引物对收集的gdna样品进行不对称pcr扩增,以通过mlpa诊断的sma病人的gdna为模板,同时以非sma患者的gdna为模板作为对照,扩增体系如下:
热循环条件为图2所示。
3.crispr/cas14a1切割荧光检测
检测切割体系如下:
将上述体系内除fq-probe和template外的其余组分加至一个干净的1.5ml离心管中,37℃孵育10分钟,然后再将fq-probe和template加入至体系中,混匀,荧光仪或者qpcr仪检测读取初始荧光值,37℃孵育60分钟再次读取荧光值,最终得到荧光增加值。
结果显示sma患者组与非患者组荧光增加值有显著性差异(图3)。在sma患者中,不对称pcr扩增出的ssdna中没有smn1外显子7扩增子激活cas14a1,ssdna探针不会被切割,因而切割产生的荧光信号比较低。相反,正常个体中可以扩增出smn1外显子7,产生强烈的荧光信号。因此,实验证实本发明可有效的检查出sma患者。
实施例3
对比不同长度的fq-probe影响
我们构建了12nt与21nt长度的fq-probe,以正常人gdna扩增产物为模板,进行crispr/cas14a1切割荧光检测,具体步骤与实施例2类似。结果显示,21nt长度的fq-probe产生的δ荧光值明显高于12nt的fq-probe产生的δ荧光值(图4),因此我们采用21nt长度的fq-probe。
实施例4
与硫代磷酸化修饰的pcr扩增对比
参照2018年doudna等报道的技术方案,我们同时利用硫代磷酸化修饰的pcr扩增引物f与普通的pcr引物r进行扩增之后,加入t7核酸外切酶制备ssdna,再进行crispr/cas14a1切割检测,结果显示,sma与正常人的荧光值也是有明显区别的,但是荧光值差只有2000左右(图5),但是与图3中荧光值差20000多相比,sma与正常人δ荧光值相差较小,仅有图3的10%。而在此次试验中,用的初始gdna量是2ng/ul,结果是δ荧光值相差较小,在初始gdna模板量更低的情况下,会造成δ荧光值相差更小,可能不能区分开sma与正常人。因此,相比于与硫代磷酸化修饰的pcr扩增,本发明的系统联合不对称pcr检测能大幅提高检测准确性。
实施例5
检测下限
针对sma病人gdna与非病人gdna进行浓度测定,并对gdna进行梯度稀释,分别以2ng/μl,1ng/μl,0.5ng/μl,0.1ng/μl,0.05ng/μl,0.01ng/μl,0.005ng/μl,0.001ng/μl浓度的sma病人gdna与非病人gdna进行不对称pcr扩增,扩增产物进行crispr/cas14a1切割反应检测。在不同浓度下,sma患者与非sma患者,不对称pcr扩增出的ssdna对cas14a1激活能力不同,产生的荧光信号不同,不同浓度gdna,比较sma患者与非患者的荧光增加值,显示在0.01ng/μl浓度时,sma患者与非患者组荧光增加值依然有显著性差异,但在0.005ng/μl,0.001ng/μl浓度,sma患者与非患者组荧光增加值没有显著性差异(图6),因此该方法的检测下限为0.01ng/μl,按照人类基因组的分子量(1.9×1012g/mol,~2.9gb)换算,该方法检测下限约为5.26amol/l。
实施例6
临床评估灵敏度与特异性
对临床上收集的33例样本dna进行不对称pcr扩增,扩增产物经crispr/cas14a1切割荧光检测,通过荧光增加值判定是否sma患者,如图7所示,为样本的荧光检测结果,判定结果与mlpa检测结果比对,如表1所示,使用本研究中检测方法检测结果与mlpa检测结果均一致,显示这种检测方法可以准确检测出sma患者,其灵敏度和特异性均达到100%。
表1crispr/cas14a1检测与mlpa检测临床样本结果对照
上述实施例阐明的内容应当理解为这些实施例仅用于更清楚地说明本发明,而不用于限制本发明的范围,在阅读了本发明之后,本领域技术人员对本发明的各种等价形式的修改均落入本申请所附权利要求所限定的范围。
sequencelisting
<110>中南大学
<120>一种诊断脊髓性肌萎缩症的crispr-cas系统及其应用
<130>1
<160>4
<170>patentinversion3.5
<210>1
<211>200
<212>dna
<213>人工合成
<400>1
taatacgactcactataggttcactgataaagtggagaaccgcttcaccaaaagctgtcc60
cttaggggattagaacttgagtgaaggtgggctgcttgcatcagcctaatgtcgagaagt120
gctttcttcggaaagtaaccctcgaaacaaattcatttgaaagaatgaaggaatgcaaca180
ttttgtctgaaaccctgtaa200
<210>2
<211>25
<212>dna
<213>人工合成
<400>2
aaataagtaaaatgtcttgtgaaac25
<210>3
<211>22
<212>dna
<213>人工合成
<400>3
gaatgtgagcaccttccttctt22
<210>4
<211>21
<212>dna
<213>人工合成
<400>4
ttttttttttttttttttttt21
- 还没有人留言评论。精彩留言会获得点赞!