新型反应型苯并三唑紫外线吸收剂及其用途的制作方法
本发明涉及一种新型具有红位移的苯并三唑紫外线吸收剂及其用途,尤其涉及一种具有优异的紫外线吸收能力的苯并三唑紫外线吸收剂、及包含其的组合物、眼镜镜片及保护贴。
背景技术:
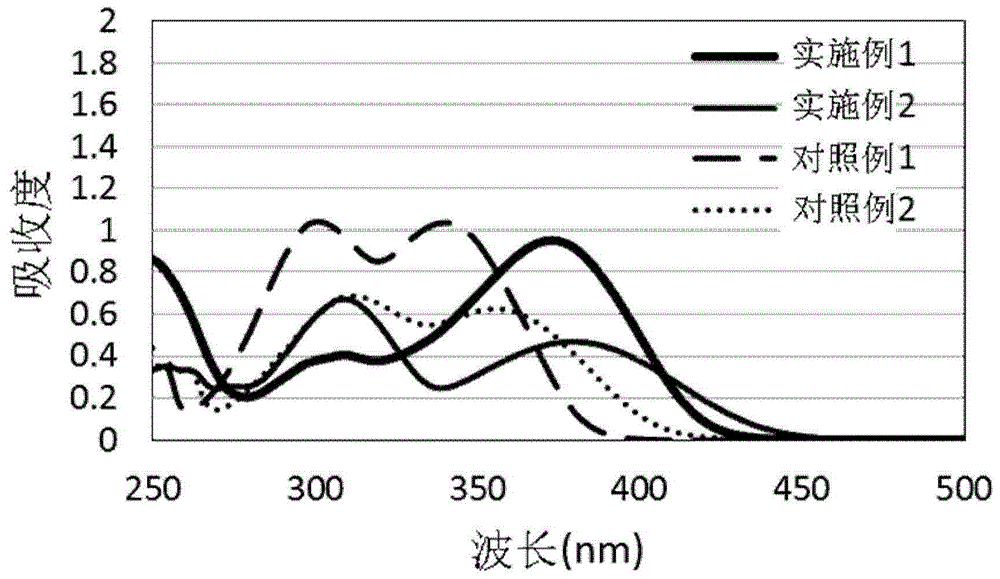
:紫外线吸收剂为一种光稳定剂,其能吸收阳光或荧光光源中的紫外线,进而保护添加有紫外线吸收剂的物质不受到紫外线的破坏,或者是,当含有紫外线吸收剂的涂层涂布于基材上时,也能保护基材不受到紫外线的破坏。目前,已知的紫外线吸收剂主要可分为苯并三唑类、水杨酸酯类、苯酮类、取代丙烯腈类和三嗪类。其中,苯并三唑紫外线吸收剂为一具有高稳定性的紫外线吸收剂,而可有效帮助包含有苯并三唑紫外线吸收剂的物质或基材对抗紫外线的破坏。然而,市场上现有苯并三唑紫外线吸收剂仍有其吸收极限,约400nm。有鉴于此,目前仍需发展一种新颖的苯并三唑紫外线吸收剂,其吸收光谱可具有红位移的效果,进而使苯并三唑紫外线吸收剂的应用领域更加广泛。技术实现要素:本发明的主要目的在于提供一种新颖化合物,其具有优异的紫外线吸收能力及抗萃取特性。本发明提供的化合物如下式(i)所示:其中,a为-s-或-so2-;r1为c1至c10的直链或支链亚烷基、含羟基取代的c1至c10的直链或支链亚烷基、或被酯基中断的c1至c10的直链或支链亚烷基;r2为-r3y、氢、c1至c10的直链或支链烷基、或c6至c15芳烷基;r3为c1至c10的直链或支链亚烷基、含羟基取代的c1至c10的直链或支链亚烷基、或被酯基中断的c1至c10的直链或支链亚烷基;x及y各自独立为-oh、-ocor4、-nh2、-ncor4、-nco、-co2h、-co2r4或r4为c1至c10的直链或支链烷基、或c3至c10的直链或支链烯基;r5为c3至c10的亚环烯基;以及r6为c1至c10的直链或支链亚烷基或1,2-亚苯基。本发明所提供的新颖化合物为一苯并三唑化合物,其可作为一紫外线吸收剂。其中,在式(i)中的苯并环上5号位置基团为含硫原子的基团,而在式(i)中的苯环上5号位置基团为含氧原子的基团;在此两种基团的协同作用下,可使其吸收光谱具有红位移的效果,而具有广域的吸收范围。特别是,本发明所提供的新颖化合物其吸收范围可延伸至450nm之后,并在280nm至340nm波段中保有已知现有技术的常规紫外线吸收剂的吸收波峰;借此,可应用于抗蓝光的材料,如镜片、隐形眼镜等产品,还可应用于航太及车辆所用的碳纤复合材料。此外,本发明所提供的新颖化合物还通过引入一反应性基团,可以与高分子单体或寡聚物进行共聚生成含有紫外线吸收剂的高分子材料,并且所得的高分子材料中的紫外线吸收剂有良好的抗萃取特性。在本发明中,a可为-s-或-so2-。在本发明中,r1可为c1至c10的直链或支链亚烷基、含羟基取代的c1至c10的直链或支链亚烷基、或被酯基中断的c1至c10的直链或支链亚烷基。在本发明的一实施例中,r1可为c1至c6的直链或支链亚烷基、含羟基取代的c1至c6的直链或支链亚烷基、或被酯基中断的c1至c6的直链或支链亚烷基。在本发明的另一实施例中,r1可为c1至c3的直链或支链亚烷基、含羟基取代的c1至c3的直链或支链亚烷基、或被酯基中断的c1至c3的直链或支链亚烷基。在本发明中,r2可为-r3y、氢、c1至c10的直链或支链烷基、或c6至c15芳烷基。在本发明的一实施例中,r2可为-r3y、氢、c1至c6的直链或支链烷基、或苄基。在本发明的另一实施例中,r2可为-r3y、氢、c1至c4的直链或支链烷基、或苄基。在本发明中,r3可为c1至c10的直链或支链亚烷基、含羟基取代的c1至c10的直链或支链亚烷基、或被酯基中断的c1至c10的直链或支链亚烷基。在本发明的一实施例中,r3可为c1至c6的直链或支链亚烷基、含羟基取代的c1至c6的直链或支链亚烷基、或被酯基中断的c1至c6的直链或支链亚烷基。在本发明的另一实施例中,r3可为c1至c3的直链或支链亚烷基、含羟基取代的c1至c3的直链或支链亚烷基、或被酯基中断的c1至c3的直链或支链亚烷基。在本发明中,x及y可各自独立为-oh、-ocor4、-nh2、-ncor4、-nco、-co2h、-co2r4或其中r4可为c1至c10的直链或支链烷基、或c3至c10的直链或支链烯基;r5可为c3至c10的亚环烯基;而r6可为c1至c10的直链或支链亚烷基或1,2-亚苯基。在本发明的一实施例中,x及y可各自独立为-oh、-ocor4、-nh2、-co2r4或r4可为c1至c6的直链或支链烷基、或c3至c6的直链或支链烯基;r5可为c3至c10的亚环烯基;而r6可为c1至c6的直链或支链亚烷基。在本发明的另一实施例中,x及y可各自独立为-oh、-ocor4a、-nh2、-co2r4b或r4a可为c3至c6的直链或支链烯基;而r4b可为c1至c6的直链或支链烷基。在本发明的一实施例中,a可为-s-;r1可为c1至c10的直链或支链亚烷基、或含羟基取代的c1至c10的直链或支链亚烷基;x可为-oh、或-co2r4;r4可为c1至c10的直链或支链烷基;而r5可为c3至c10的亚环烯基。在本发明的另一实施例中,a可为-s-;r1可为c1至c6的直链或支链亚烷基、或含羟基取代的c1至c6的直链或支链亚烷基;x可为-oh、或-co2r4;r4可为c1至c6的直链或支链烷基。在本发明的另一实施例中,a可为-s-;r1可为c1至c4的直链或支链亚烷基、或含羟基取代的c1至c4的直链或支链亚烷基;x可为-oh、或-co2r4;r4可为c1至c4的直链或支链烷基。在本发明的一实施例中,a可为-so2-;r1可为c1至c10的直链或支链亚烷基、或被酯基中断的c1至c10的直链或支链亚烷基;x可为-oh、-ocor4、-nh2或r4可为c3至c10的直链或支链烯基;而r6可为c1至c10的直链或支链亚烷基。在本发明的另一实施例中,a可为-so2-;r1可为c1至c6的直链或支链亚烷基、或被酯基中断的c1至c6的直链或支链亚烷基;x可为-oh、-ocor4、-nh2或r4可为c3至c6的直链或支链烯基。在本发明的另一实施例中,a可为-so2-;r1可为c1至c4的直链或支链亚烷基、或被酯基中断的c1至c4的直链或支链亚烷基;x可为-oh、-ocor4、-nh2或r4可为c3至c4的直链或支链烯基。在本发明的一实施例中,r2可为氢、c1至c10的直链或支链烷基、或c6至c15芳烷基。在本发明的另一实施例中,r2可为氢、c1至c6的直链或支链烷基、或苄基。在本发明的另一实施例中,r2可为氢、c1至c4的直链或支链烷基、或苄基。在本发明的一实施例中,r2可为-r3y;r3可为c1至c10的直链或支链亚烷基、或被酯基中断的c1至c10的直链或支链亚烷基;y可为-oh、-ocor4、-nh2、r4可为c3至c10的直链或支链烯基;r5可为c3至c10的亚环烯基;而r6为c1至c10的直链或支链亚烷基。在本发明的另一实施例中,r2可为-r3y;r3可为c1至c6的直链或支链亚烷基、或被酯基中断的c1至c6的直链或支链亚烷基;y可为-oh、-ocor4、-nh2、r4可为c3至c6的直链或支链烯基。在本发明的另一实施例中,r2可为-r3y;r3可为c1至c3的直链或支链亚烷基、或被酯基中断的c1至c3的直链或支链亚烷基;y可为-oh、-ocor4、-nh2、r4可为c3至c4的直链或支链烯基。本发明所提供的化合物可为下式(i-1)至(i-13)的任一化合物:在本发明中,所谓的“(亚)烷基”包括直链及支链的(亚)烷基,例如,包括直链及支链的c1至c10(亚)烷基、c1至c6(亚)烷基、或c1至c4(亚)烷基;且其具体例子包括,但不限于:(亚)甲基、(亚)乙基、(亚)丙基、(亚)异丙基、(亚)丁基、(亚)异丁基、(亚)仲丁基、(亚)叔丁基、(亚)戊基、(亚)新戊基及(亚)己基。在本发明中,所谓的“烯基”一词指包含至少一个双键且包括直链及支链的碳氢基团,例如,包含至少一个双键且包括直链及支链的c3至c10碳氢基团、c3至c6碳氢基团、或c3至c4碳氢基团;且其具体例子包括,但不限于:乙烯、丙烯及丁烯。在本发明中,所谓的“(亚)环烯基”一词指单价或双价不饱和环状碳氢基团,其包含,例如,一或以上双键及3至10个碳原子(如c3至c10)、5至8个碳原子(c5至c8)或5至7个碳原子(c5至c7);且其具体例子包含,但不限于:(亚)环戊烯基、(亚)环己烯基及(亚)环庚烯基。在本发明中,所谓的“芳基”包括6元碳单环、10元碳双环、14元三环芳香族环系统;且其具体例子包括,但不限于:苯基、萘基、芘基、蒽基及菲基。在本发明中,所谓的“芳烷基”包括本发明中所定义的烷基经加上至少一芳基所形成的分子基团。在本发明中,所谓的“被酯基中断的亚烷基”包括本发明中所定义的亚烷基的相邻两个碳原子间插入一酯基、或是亚烷基的一端连接一酯基。在本发明的一实施例中,“被酯基中断的亚烷基”可为-c(=o)o-(亚烷基)。此外,本发明还提供一种对于光引发的降解具有安定性的组合物,包含:(a)受到光引发会降解的有机材料;以及(b)前述本发明的新颖化合物。其中,以有机材料为基准,本发明的新颖化合物的含量可为有机材料的0.1%至30%。在本发明的一实施例中,前述组合物是用于形成一涂层。特别是,该涂层是形成于一对波长大于380nm的电磁辐射敏感的基材上。其中,基材的种类并无特殊限制,可为玻璃、塑胶、聚合物、硅水胶、树脂、碳纤复合材料或其混合材料。再者,本发明还提供一种具有抗蓝光效果的眼镜镜片,包含前述本发明的新颖化合物;其中,本发明的新颖化合物是涂布于眼镜镜片用的基材上,以形成一抗紫外光或抗蓝光涂层。此外,本发明还提供一种具有抗蓝光效果的保护贴,包含前述本发明的新颖化合物;其中,本发明的新颖化合物是涂布于保护贴用的基材上,以形成一抗紫外光或抗蓝光涂层。附图说明图1为实施例1、2及对照例1、2的uv吸收光谱图;图2为实施例2、3、8的uv吸收光谱图;图3为实施例2及对照例1的穿透率结果图。具体实施方式以下通过具体实施例说明本发明的实施方式,本领域技术人员可由本说明书所公开的内容轻易地了解本发明的其他优点与功效。本发明也可通过其他不同的具体实施例加以施行或应用,本说明书中的各项细节也可针对不同观点与应用,在不悖离本发明的精神下进行各种修饰与变更。除非文中另有说明,否则说明书及所附权利要求书中所使用的术语“或”通常包括“及/或”的含义。本发明将通过实施例更具体地说明,但该些实施例并非用于限制本发明的保护范围。除非特别指明,在下列实施例与比较例中用于表示成分含量以及物质量的“%”以重量为基准。制备例1:原料化合物1a的合成制备化合物1a’将856.8克叔丁基氢醌溶于2.5升甲醇中,向该溶液中加入206.2克氢氧化钠、1升水、482.3克氯丙醇和6.3克碘化钾,在氮气下回流温度加热48小时,反应混合物冷却至室温,加入5.6升水稀释,以4升二氯甲烷萃取,合并的二氯甲烷层用水洗涤,以无水硫酸钠干燥并浓缩,将残留物进行减压蒸馏,馏出物以甲苯再结晶进一步纯化,得到396.0克白色固体,熔点78.5-80.5℃。制备化合物1a步骤(a):将90.6克4-氯-2-硝基苯胺和180毫升浓盐酸在室温下搅拌一小时,并以160毫升水和300克冰稀释,在-5℃下,将37.7克亚硝酸钠溶在140毫升水中后滴加到混合物中,入毕在0℃下搅拌1小时,加入氨基磺酸直到碘化钾淀粉测试结果为阴性,接着过滤混合物,该滤液在-5℃至0℃下滴加至112.2克原料化合物1a、60克氢氧化钠和2升水的混和溶液中搅拌,当加入约1/3的重氮溶液后,将400毫升的10%氢氧化钠水溶液一起滴加到反应混合物中,两种滴加在同一时间结束。在滴加过程中,反应混合物均保持在0℃以下,入毕同温下搅拌2小时,然后回至室温。偶氮染料以盐酸酸化分离,过滤并用水洗涤,不经进一步纯化即可用于下一步反应。步骤(b):将上一步的偶氮染料溶于1.5升乙醇中,在室温氮气下,向该溶液中缓慢加入葡萄糖溶液(180克配制在1.5升2n氢氧化钠水溶液中),过程中反应温度保持在30℃以下,以薄层层析法追踪至反应完成。然后加入165克新鲜活化的锌粉,该混合物在室温下搅拌3小时后,以1升水稀释并搅拌15分钟,静置1小时,过滤分离沉淀物后,先以水洗涤,再以2升热乙醇萃取滤饼,直到滤饼只剩下锌残留。将萃取液冷却至室温,过滤分离固体,再以冷乙醇洗涤,真空干燥后得到67.8克黄色固体,熔点134.7℃。1hnmr(400mhz,cdcl3):δ11.35(1h),7.92(1h),7.87(1h),7.80(1h),7.43(1h),7.02(1h),4.20(2h),3.91(2h),2.10(2h),1.80(1h),1.49(9h);tga(5%weightloss):246.23℃。实施例1:化合物2a的合成(即本发明式(i-1)化合物)在20毫升反应瓶中,置入0.75克化合物2a、0.52克氢氧化钾、0.09克碘化钾、0.5毫升2-巯基乙醇、2毫升n-甲基吡咯烷酮,加热至100℃后搅拌12小时,冷却回到室温后以1n盐酸水溶液酸化至ph5。混合液以100毫升甲苯、50毫升乙酸乙酯、100毫升水萃取,排去水层后有机层以硫酸镁干燥除水,减压浓缩后以管柱纯化获得0.21克化合物2a,产率25.2%。1hmr(cdcl3,400mhz):1.49(s,9h),1.86(s,1h),2.10(quint,j=5.9hz,2h),3.25(t,j=5.8hz,2h),3.80-3.95(m,4h),4.20(t,j=6.0hz,2h),7.06(d,j=3.0hz,1h),7.42(dd,j=1.4,8.8hz,1h),7.79(d,j=3.0hz,1h),7.82(d,j=8.8hz,1h),7.83(d,j=1.4hz,1h),11.42(s,1h);13cnmr(cdcl3,100mhz):29.5,32.2,35.8,36.9,60.5,60.8,103.1,115.8,117.2,118.1,125.3,129.9,136.0,141.1,141.6,143.3,143.6,151.2;hrms(esi+,m+h):calc.418.1801,found418.1804。实施例2:化合物2b的合成(即本发明式(i-2)化合物)在250毫升反应瓶中,置入0.75克化合物2a、0.1克na2wo4·2h2o、9毫升90%甲酸水溶液、50毫升甲苯、9毫升30%过氧化氢水溶液后加热至70℃后搅拌6小时,冷却回到室温后混合液以100毫升甲苯、10毫升水进行萃取。水层再以100毫升甲苯萃取后合并有机层,有机层经100毫升饱和食盐水清洗、硫酸镁干燥、减压浓缩与管柱纯化获得0.23克化合物2b,产率28.3%。1hnmr(cdcl3,400mhz):1.49(s,9h),2.04-2.17(m,2h),2.80(s,1h),3.47(dt,j=6.0,2.4hz,2h),3.92(t,j=6.0hz,2h),4.09(t,j=5.2hz,2h),4.21(t,j=6.0hz,2h),7.07(d,j=3.2hz,1h),7.83(d,j=3.2hz,1h),7.94(d,j=9.1hz,1h),8.12(dd,j=0.8,9.1hz,1h),8.66(d,j=0.8hz,1h),11.22(s,1h);13cnmr(cdcl3,100mhz):29.5,32.2,35.9,56.5,58.6,60.5,66.5,103.2,118.6,119.5,120.8,125.0,138.3,141.5,141.7,144.1,144.4,151.4;hrms(esi+,m+h):calc.450.1699,found450.1710。实施例3:化合物2c的合成(即本发明式(i-3)化合物)在250毫升反应瓶中,置入0.85克化合物2b、0.1克对苯二酚、0.1克对甲苯磺酸、0.5毫升甲基丙烯酸与125毫升甲苯后加热至回流搅拌16小时,冷却回到室温后混合液以100毫升饱和碳酸氢钠水溶液进行萃取。水层再以50毫升甲苯萃取后合并有机层,有机层经100毫升饱和食盐水清洗、硫酸镁干燥、减压浓缩与管柱纯化后经甲醇/甲苯结晶获得0.34克化合物2c,产率30.7%。1hnmr(cdcl3,400mhz):1.50(s,9h),1.71(d,j=1.4hz,3h),1.97(d,j=1.4hz,3h),2.23(quint,j=6.2hz,2h),3.63(t,j=6.0hz,2h),4.17(t,j=6.0hz,2h),4.40(t,j=6.2hz,2h),4.56(t,j=5.8hz,2h),5.37(dd,j=0.9,1.4hz,1h),5.58(dd,j=0.9,1.4hz,1h),5.75(s,1h),6.14(s,1h),7.08(d,j=2.8hz,1h),7.81(d,j=2.8hz,1h),7.93(dd,j=1.2hz,9.2hz,1h),8.11(d,j=9.2hz,1h),8.66(d,j=1.2hz,1h),11.24(s,1h);13cnmr(cdcl3,100mhz):18.1,18.5,28.8,29.5,35.9,55.4,58.0,61.7,65.3,103.0,119.0,119.4,121.0,124.9,125.1,125.7,126.6,135.3,136.5,138.7,141.6,144.1,144.4,151.5,166.6,167.5;hrms(esi+,m+h):calc.586.2223,found586.2227。实施例4:化合物2d的合成(即本发明式(i-4)化合物)在25毫升反应瓶中,置入0.3克化合物1a、2.0克碳酸钾、1.2克2-胺基乙硫醇与2毫升n-甲基吡咯烷酮后加热至100℃搅拌12小时,冷却回到室温后混合液倒入100毫升水中并以1nhcl水溶液酸化至ph5。混合液加入25毫升异丙醇、75毫升甲苯与10克氯化钠后进行萃取。水层以25毫升异丙醇与75毫升甲苯萃取后合并有机层,有机层经100毫升饱和食盐水清洗、减压浓缩与管柱纯化获得中间体3a,全数用于后续反应。1hnmr(pyridine-d5,400mhz):1.59(s,9h),2.29(quint,j=6.2hz,2h),3.82(t,j=7.1hz,2h),4.05(t,j=7.1hz,2h),4.13(t,j=6.2hz,2h),4.37(t,j=6.2hz,2h),7.24(d,j=2.6hz,1h),7.55(d,j=9.0hz,1h),7.90(d,j=9.0hz,1h),8.00(d,j=2.6hz,1h),8.19(s,1h);hrms(esi+,m+h):calc.417.1960,found417.1971。中间体3a加入30毫升乙酸乙酯、30毫升叔丁醇、3毫升醋酸酐与3毫升三乙胺后加热至70℃搅拌待中间体3a全溶后降温回到室温。反应液加入100毫升水与100毫升乙酸乙酯进行萃取,水层再以50毫升乙酸乙酯进行萃取后合并有机层,有机层经50毫升饱和食盐水清洗与减压浓缩获得中间体3b,全数用于后续反应。中间体3b加入0.1克na2wo4·2h2o、15毫升30%过氧化氢水溶液、5毫升水与50毫升异丙醇后室温下搅拌16小时。反应液加入100毫升水与100毫升乙酸乙酯进行萃取,水层再以50毫升乙酸乙酯进行萃取后合并有机层,有机层经100毫升饱和食盐水清洗与减压浓缩获得中间体3c,全数用于后续反应。hrms(esi+,m+h):calc.533.2070,found533.2074。中间体3c加入85%浓磷酸3毫升与1毫升水搅拌加热回流8小时后降温回到室温。反应液倒入100毫升水中,以45%naoh水溶液中和至ph6.5。混合液加入100毫升异丙醇、100毫升甲苯与10克氯化钠后进行萃取。水层以50毫升异丙醇与50毫升甲苯萃取后合并有机层,有机层经100毫升饱和食盐水清洗、减压浓缩与甲苯/甲醇结晶获得0.12克化合物2d,4步总产率33.5%。1hnmr(dmso-d6,400mhz):1.45(s,9h),1.89(quint,j=6.3hz,2h),2.94(s,2h),3.59(t,j=6.3hz,2h),3.65(s,2h),4.06(t,j=6.3hz,2h),7.00(d,j=2.8hz,1h),7.49(t,j=2.8hz,1h),7.98(d,j=8.8hz,1h),8.35(d,j=8.8hz,1h),8.72(d,j=2.8hz,1h);13cnmr(dmso-d6,100mhz):29.2,32.1,34.7,35.3,55.3,57.3,65.3,104.9,117.3,119.9,120.5,124.8,126.4,137.9,140.6,141.9,143.0,144.6,151.1;hrms(esi+,m+h):calc.449.1859,found449.1859。制备例2:原料化合物1b的合成前驱物化合物4的合成在250毫升反应瓶中,置入33.24克叔丁基对苯二酚与20.42克2-甲基-2-唑啉后加热至160℃搅拌反应16小时。反应液冷却回到室温后以100毫升甲苯、200毫升乙酸乙酯与100毫升水进行萃取,排除水层后有机层再以100毫升饱和食盐水清洗。有机层干燥浓缩后经减压蒸馏收集压力0.25mbar沸点215-220℃区间馏出液获得30.31克化合物4,产率42.2%。1hnmr(acetone-d6,400mhz):1.38(s,9h),1.93(s,3h),3.53(q,j=5.6hz,2h),3.94(t,j=5.6hz,2h),6.58(dd,j=2.8,8.8hz,1h),6.74(d,j=8.8hz,1h),6.78(d,j=2.8hz,1h),7.51(brs,1h),8.18(brs,1h);13cnmr(acetone-d6,100mhz):22.8,29.8,35.2,39.8,67.8,112.2,115.0,117.2,137.6,150.8,152.7,170.8。原料化合物1b的合成将16克4-氯-2-硝基苯胺、0.05克分散剂、50毫升浓盐酸与100克冰混合均匀后搅拌1小时。混合液降温至5℃后缓慢加入亚硝酸纳水溶液(6.8克亚硝酸钠溶于50毫升水中),入毕后继续搅拌0.5小时,再加入0.3克氨基磺酸铵搅拌0.5小时。最后加入0.3克硅藻土搅拌0.5小时,过滤混合液得到偶氮液,保存在低温(<5℃)用于后续反应步骤。取18.71克化合物4、分散剂0.1克、45%氢氧化钠水溶液10克、冰100克与乙醇500毫升混合均匀并搅拌1小时。降温至5℃后缓慢滴加上述制备的偶氮液,在适当时后加入45%氢氧化钠水溶液控制ph值维持8到10.5间。滴加完毕后回到室温并搅拌16小时,再加入66克45%氢氧化钠水溶液后将反应液加热至回流并搅拌0.5小时。加入16克连二亚硫酸钠并搅拌1小时,再加入16克连二亚硫酸钠并搅拌1小时,再追加32克连二亚硫酸钠与16克45%氢氧化钠水溶液并搅拌1小时后降温回到室温,过滤收集固体获得13.3克化合物1b,产率44.3%。1hnmr(dmso-d6,400mhz):1.45(s,9h),1.84(s,3h),3.43(q,j=5.2hz,2h),4.02(t,j=5.2hz,2h),7.00(s,1h),7.53(s,1h),7.59(d,j=9.0hz,1h),8.12(s,1h),8.14(d,j=9.0hz,1h),8.25(s,1h),10.64(brs,1h);13cnmr(dmso-d6,100mhz):22.5,29.2,35.3,38.3,67.1,104.6,116.9,119.7,126.0,129.0,132.4,140.4,141.4,142.8,143.2,150.7,169.5。实施例5:化合物2e的合成(即本发明式(i-5)化合物)在25毫升反应瓶中,置入1.25克化合物1b、4.0克碳酸钾、1.2克2-胺基乙硫醇与10毫升n-甲基吡咯烷酮后加热至100℃搅拌12小时,冷却回到室温后混合液倒入300毫升水中并以1nhcl水溶液酸化至ph6。混合液加入25毫升异丙醇、75毫升甲苯与20克氯化钠后进行萃取。水层以25毫升异丙醇与75毫升甲苯萃取后合并有机层,有机层经100毫升饱和食盐水清洗、减压浓缩与管柱纯化获得中间体3d,全数用于后续反应。中间体3d加入50毫升乙酸乙酯、9毫升叔丁醇、12毫升醋酸酐与9毫升三乙胺后加热至70℃搅拌待中间体3d全溶后降温回到室温。反应液加入100毫升水与100毫升乙酸乙酯进行萃取,水层再以50毫升乙酸乙酯进行萃取后合并有机层,有机层经50毫升饱和食盐水清洗与减压浓缩获得中间体3e,全数用于后续反应。中间体3e加入0.1克na2wo4·2h2o、15毫升30%过氧化氢水溶液、5毫升水与50毫升异丙醇后室温下搅拌16小时。反应液加入100毫升水与100毫升乙酸乙酯进行萃取,水层再以50毫升乙酸乙酯进行萃取后合并有机层,有机层经100毫升饱和食盐水清洗与减压浓缩获得中间体3f,全数用于后续反应。中间体3f加入85%浓磷酸12毫升与3毫升水搅拌加热回流8小时后降温回到室温。反应液倒入200毫升水中,以45%naoh水溶液中和至ph6.5,过滤收集固体,并以50毫升水清洗表面。滤饼以异丙醇再结晶后获得0.13克化合物2e,4步总产率9.7%。1hnmr(dmso-d6,400mhz):1.46(s,9h),2.92(t,j=7.2hz,2h),3.18(t,j=4.8hz,2h),3.64(t,j=7.2hz,2h),4.22(t,j=4.8hz,2h),7.11(d,j=2.8hz,1h),7.55(d,j=2.8hz,1h),7.98(d,j=2.8hz,1h),8.00(dd,j=1.2,9.2hz,1h),8.38(d,j=9.2hz,1h),8.74(d,j=1.2hz,1h);13cnmr(dmso-d6,100mhz):29.2,35.2,35.3,38.6,56.0,65.7,105.8,117.7,119.9,120.5,124.8,126.5,138.1,140.7,142.0,143.6,144.7,150.3;hrms(esi+,m+h):calc.434.1862,found434.1863。实施例6:化合物2f的合成(即本发明式(i-6)化合物)在25毫升反应瓶中,置入2.88克化合物1b、3.0克碳酸钾、3.0克2-胺基乙硫醇与10毫升n-甲基吡咯烷酮后加热至100℃搅拌12小时,冷却回到室温后混合液倒入300毫升水中并以1nhcl水溶液酸化至ph5。过滤收集固体并以100毫升水清洗表面获得1.5克中间体3d,全数用于后续反应。中间体3d加入85%浓磷酸12毫升与3毫升水搅拌加热回流8小时后降温回到室温。反应液倒入200毫升水中,以45%naoh水溶液中和至ph6.5,过滤收集固体,并以50毫升水清洗表面。滤饼以异丙醇再结晶后获得0.5克中间体3g。取0.15克中间体3g加入0.15克降冰片烯二酸酐与20毫升甲苯搅拌回流16小时,降温到室温后经减压浓缩与管柱层析后获得0.1克化合物2f,3步总产率15.1%。1hnmr(cdcl3,400mhz):1.47(dt,j=8.8,1.6hz,1h),1.48(s,9h),1.54(dt,j=8.8,1.6hz,1h),1.73(dt,j=8.8,1.6hz,1h),1.74(dt,j=8.8,1.6hz,1h),3.10(t,j=7.6hz,2h),3.24-3.27(m,1h),3.29-3.32(m,1h),3.38-3.43(m,4h),3.66(t,j=7.6hz,2h),3.81(t,j=5.6hz,2h),4.10(t,j=5.6hz,2h),6.08(t,j=1.6hz,2h),6.14(t,j=1.6hz,2h),6.95(d,j=3.0hz,1h),7.40(dd,j=8.8,3.2hz,1h),7.72(d,j=3.0hz,1h),7.83(d,j=8.8hz,1h),7.90(d,j=3.2hz,1h),11.46(s,1h);13cnmr(cdcl3,100mhz):29.5,30.3,35.7,37.3,37.8,45.4,45.9,52.2,52.3,64.9,103.2,115.3,117.0,118.0,125.2,129.5,134.4,134.6,135.7,140.9,141.5,143.3,143.6,150.7,177.5,177.6;hrms(esi+,m+h):calc.694.2699,found694.2775。制备例3:原料化合物1c的合成原料化合物1c’的合成制备方法参照化合物1a’的合成,起始物为856.8克的叔丁基氢醌和960.0克的溴化苄,得到670.0克淡黄色液体。原料化合物1c的合成制备方法如化合物1a的合成,起始物改为450.0克的原料化合物1c’,得到138.0克黄色固体,熔点130.7℃。1hnmr(400mhz,cdcl3):δ11.36(1h),7.80-8.00(3h),7.49(2h),7.30-7.45(4h),7.10(1h),5.11(2h),1.48(9h);hrmsesi[m-h]-:calc.406.1322,found406.1324。实施例7:化合物2g的合成(即本发明式(i-7)化合物)在20毫升反应瓶中,置入2.0克化合物1c、0.6克氢氧化钾、10.0克碳酸钾、3毫升2-巯基乙醇、20毫升n-甲基吡咯烷酮,加热至140℃后搅拌8小时,冷却回到室温后以1n盐酸水溶液酸化至ph5。混合液以100毫升甲苯、50毫升乙酸乙酯、100毫升水萃取,排去水层后有机层以硫酸镁干燥除水,减压浓缩后以管柱纯化获得0.8克化合物2g,产率36.3%。1hnmr(cdcl3,400mhz):1.48(s,9h),3.25(t,j=6.0hz,2h),3.87(t,j=6.0hz,2h),5.12(s,2h),7.09(d,j=3.2hz,1h),7.29-7.55(m,6h),7.77-7.93(m,3h),11.44(s,1h);13cnmr(cdcl3,100mhz):29.6,35.8,36.9,60.5,70.9,103.4,115.9,117.6,118.1,125.3,127.9,128.2,128.8,129.9,136.0,137.0,141.1,141.6,143.3,143.7,151.3;hrms(esi+,m+h):calc.450.1851,found450.1863。实施例8:化合物2h的合成(即本发明式(i-8)化合物)在250毫升反应瓶中,置入0.4克化合物2g、0.1克na2wo4·2h2o、15毫升30%过氧化氢水溶液、50毫升异丙醇与5毫升水后搅拌16小时,混合液加入100毫升甲苯与10克氯化钠进行萃取。水层再以100毫升甲苯萃取后合并有机层,有机层经100毫升饱和食盐水清洗、硫酸镁干燥、减压浓缩与管柱纯化获得0.28克化合物2h,产率65.3%。1hnmr(cdcl3,400mhz):1.49(s,9h),2.66(t,j=2.4hz,1h),3.47(dt,j=2.4,5.2hz,2h),4.09(t,j=5.2hz,2h),5.14(s,2h),7.16(d,j=2.8hz,1h),7.30-7.47(m,3h),7.50(d,j=7.2hz,2h),7.89-7.99(m,2h),8.14(d,j=9.2hz,1h),8.68(s,1h),11.25(s,1h);13cnmr(cdcl3,100mhz):29.5,35.9,56.6,58.6,71.0,103.5,119.0,119.6,120.9,125.0,127.9,128.3,128.8,136.8,138.3,141.6,141.7,144.3,144.5,151.4;hrms(esi+,m+h):calc.482.1750,found482.1764。实施例9:化合物2i的合成(即本发明式(i-9)化合物)在250毫升反应瓶中,置入0.2克化合物2h、0.1克对苯二酚、0.1克对甲苯磺酸、0.5毫升甲基丙烯酸与75毫升甲苯后加热至回流搅拌16小时,冷却回到室温后混合液以加入25毫升甲苯与100毫升饱和碳酸氢钠水溶液进行萃取。水层再以50毫升甲苯萃取后合并有机层,有机层经100毫升饱和食盐水清洗、硫酸镁干燥、减压浓缩与管柱纯化后经甲醇/甲苯结晶获得0.15克化合物2i,产率78.6%。1hnmr(cdcl3,400mhz):1.49(s,9h),1.72(d,j=1.6hz,3h),1.97(d,j=1.2hz,3h),3.64(t,j=6.0hz,2h),4.57(t,j=6.0hz,2h),5.38(d,j=1.6hz,1h),5.76(s,1h),7.02(d,j=2.8hz,1h),7.78(d,j=2.8hz,1h),7.92(dd,j=1.2hz,9.2hz,1h),8.08(d,j=9.2hz,1h),8.64(d,j=1.2hz,1h),11.13(s,1h);13cnmr(cdcl3,100mhz):18.1,29.5,35.9,55.4,58.0,105.6,117.9,119.5,121.1,125.1,126.8,135.3,138.6,141.6,141.7,143.8,144.4,148.1,166.7;hrms(esi+,m+h):calc.460.1542,found460.1549。制备例4:原料化合物1d的合成原料化合物1d’的合成制备方法如化合物1a’的合成,起始物为856.8克叔丁基氢醌和706.3克溴丁烷,得到590.1克淡黄色液体。原料化合物1d的合成制备方法如化合物1a的合成,起始物改为450.0克的原料化合物1d’,得到159.6克黄色固体,熔点101.6℃。hrmsesi[m-h]-:calc.372.1479,found372.1477;1hnmr(400mhz,cdcl3):δ11.31(1h),7.91(1h),7.85(1h),7.75(1h),7.41(1h),7.02(1h),4.03(2h),1.81(2h),1.54(2h),1.49(9h),1.00(3h)。实施例10:化合物2j的合成(即本发明式(i-10)化合物)在25毫升反应瓶中,置入1.0克化合物1d、1.6克碳酸钾、1.0克2-胺基乙硫醇与10毫升n-甲基吡咯烷酮后加热至120℃搅拌16小时,冷却回到室温后混合液倒入300毫升水中并以1nhcl水溶液酸化至ph5。混合液加入200毫升甲苯与10克氯化钠后进行萃取。水层以100毫升甲苯萃取后合并有机层,有机层经100毫升饱和食盐水清洗、减压浓缩与管柱纯化获得0.3克中间体3h,全数用于后续反应。hrms(esi+,m+h):calc.415.2168,found415.2164。中间体3h加入1.0克降冰片烯二酸酐、20毫升二甲基甲酰胺与20毫升甲苯搅拌回流16小时,降温到室温后加入200毫升甲苯、10克氯化钠与100毫升饱和氯化铵水溶液进行萃取。水层再以100毫升乙酸乙酯萃取后合并有机层,有机层经100毫升饱和食盐水清洗、减压浓缩与管柱纯化后获得0.21克化合物2j,2步总产率14.0%。1hnmr(cdcl3,400mhz):1.03(t,j=7.6hz,3h),1.45-1.65(m,12h),1.70-1.90(m,3h),3.08(t,j=7.2hz,2h),3.20-3.30(m,2h),3.40-3.47(m,2h),3.65(t,j=7.2hz,2h),4.04(t,j=6.4hz,2h),6.14(s,2h),7.01(d,j=2.8hz,1h),7.39(dd,j=9.2,1.4hz,1h),7.76(d,j=2.8hz,1h),7.83(d,j=9.2hz,1h),7.90(d,j=1.4hz,1h),11.43(s,1h);13cnmr(cdcl3,100mhz):14.0,19.4,29.5,30.3,31.6,35.7,37.4,45.1,45.9,52.3,68.5,102.8,115.4,117.2,118.0,125.2,129.5,134.6,135.6,140.8,141.5,143.3,143.4,151.5,177.5;hrms(esi+,m+h):calc.561.2536,found561.2608。实施例11:化合物2k的合成(即本发明式(i-11)化合物)在25毫升反应瓶中,置入1.0克化合物1d、1.6克碳酸钾、1.0毫升巯基乙酸与10毫升n-甲基吡咯烷酮后加热至140℃搅拌16小时,再补1.6克碳酸钾继续加热16小时。冷却回到室温后混合液倒入300毫升水中并以1nhcl水溶液酸化至ph5。混合液加入25毫升异丙醇、75毫升乙酸乙酯与10克氯化钠后进行萃取。水层加入25毫升异丙醇与75毫升乙酸乙酯萃取后合并有机层,有机层经100毫升饱和食盐水清洗、减压浓缩后获得中间体3i,全数用于后续反应。中间体3i加入0.5克对甲苯磺酸、20毫升甲苯与100毫升甲醇搅拌回流16小时,降温到室温后加入200毫升甲苯、10克氯化钠与100毫升饱和碳酸氢钠水溶液进行萃取。水层再以100毫升乙酸乙酯萃取后合并有机层,有机层经100毫升饱和食盐水清洗、减压浓缩与管柱纯化后获得0.34克化合物2k,2步总产率28.7%。1hnmr(cdcl3,400mhz):1.01(t,j=7.2hz,3h),1.45-1.65(m,11h),1.75-1.85(m,2h),3.76(s,3h),3.78(s,2h),3.40-3.47(m,2h),3.65(t,j=7.2hz,2h),4.03(t,j=6.4hz,2h),7.01(d,j=3.2hz,1h),7.44(dd,j=8.8,1.4hz,1h),7.75(d,j=3.2hz,1h),7.84(d,j=8.8hz,1h),7.87(d,j=1.4hz,1h),11.43(s,1h);13cnmr(cdcl3,100mhz):14.1,19.5,29.6,31.6,35.8,36.2,52.9,68.5,102.8,116.3,117.4,118.1,125.3,129.6,135.6,141.0,141.7,143.3,143.4,151.6,169.8;hrms(esi+,m+h):calc.444.1957,found444.1957。实施例12:化合物2l的合成(即本发明式(i-12)化合物)在20毫升反应瓶中,置入5.0克化合物1a、5.0克碳酸钾、3.0毫升硫代甘油、15毫升n-甲基吡咯烷酮,加热至140℃后搅拌4小时,冷却回到室温后反应液倒入200毫升水中并以1n盐酸水溶液酸化至ph4.5。过滤收集固体,并以100毫升水清洗表面。滤饼取出后加入100毫升异丙醇加热至60℃溶解,并加入300毫升庚烷后缓慢降温,再加入200毫升水并降温至5℃。收集析出固体并经管柱纯化获得4.53克化合物2l,产率76.0%。1hnmr(cdcl3,400mhz):1.45(s,9h),1.91(quint,j=6.4hz,2h),3.06(dd,j=7.0,13.2hz,1h),3.20(dd,j=4.4,13.2hz,1h),3.40-3.55(m,2h),3.60(q,j=5.8hz,2h),3.70-3.82(m,1h),4.07(t,j=6.6hz,2h),4.60(t,j=5.2hz,1h),4.80(t,j=5.8hz,1h),5.15(d,j=5.2hz,1h),6.94(d,j=3.0hz,1h),7.48(dd,j=1.2,9.0hz,1h),7.59(d,j=3.0hz,1h),7.91(d,j=1.2hz,1h),7.98(d,j=9.0hz,1h),11.05(s,1h);13cnmr(cdcl3,100mhz):29.2,32.2,35.3,36.1,57.4,64.7,65.3,70.1,103.3,112.3,116.3,117.8,125.5,129.0,138.6,140.2,140.9,142.3,143.3,151.0;hrms(esi+,m+h):calc.448.1906,found418.1913。对照例1为市售商品82,如下式(ii)所示。对照例2:化合物6的合成在20毫升反应瓶中,置入4.0克化合物1e(其为市售商品75)、1.3克氢氧化钾、0.05克碘化钾、0.9毫升2-巯基乙醇、10毫升n-甲基吡咯烷酮,加热至100℃后搅拌6小时,冷却回到室温后以1n盐酸水溶液酸化至ph5。混合液以100毫升甲苯与100毫升水萃取,排去水层后有机层以硫酸镁干燥除水,减压浓缩后以管柱纯化获得1.94克中间体5,产率43%。hrms(esi+,m+h):calc.400.2059,found400.2059。在250毫升反应瓶中,置入0.9克化合物5、0.15克na2wo4·2h2o、25毫升甲苯、50毫升异丙醇、15毫升30%过氧化氢水溶液后搅拌16小时,混合液以85毫升甲苯、50毫升水进行萃取。水层再以75毫升甲苯萃取后合并有机层,有机层经50毫升饱和食盐水清洗、硫酸镁干燥、减压浓缩获得0.89克化合物6,产率91.6%。1hnmr(cdcl3,400mhz):1.40(s,9h),1.52(s,9h),2.45(brs,1h),3.47(t,j=5.4hz,2h),4.08(t,j=5.4hz,2h),7.50(d,j=2.4hz,1h),7.94(dd,j=8.8,1.4hz,1h),8.14(d,j=8.8hz,1h),8.31(d,j=8.8hz,1h),8.69(d,j=1.4hz,1h),11.39(s,1h);13cnmr(cdcl3,100mhz):29.7,31.6,34.8,35.9,56.5,58.6,116.6,119.6,120.9,124.8,125.0,126.7,138.1,139.2,141.7,142.4,144.5,147.3;hrms(esi+,m+h):calc.432.1957,found432.1957。对照例3为市售商品78,如下式(iii)所示。对照例4:化合物7的合成将40克化合物1d溶于10.5克n-甲基吡咯酮中,在90℃下,向该溶液中缓慢加入4.43克45%氢氧化钾水溶液,接着再滴加2.04克硫酚,入毕升温至170℃,除水24小时后降温至100℃,以75毫升二甲苯萃取,以75毫升水洗涤,以15%盐酸使水层酸化后排水层,用无水硫酸钠干燥有机层并浓缩,静置析出黄色固体。而后,将所得黄色固体溶于4.28克二甲苯中,向该溶液中加入0.038克钨酸钠、0.85克90%甲酸,升温至50℃,缓慢滴加2.54克30%双氧水,滴加时反应温度不超过85℃,以薄层层析法追踪至反应结束,加入二甲苯及水,萃取干燥浓缩后,在甲醇中再结晶,得到35.4克黄色固体化合物7,熔点148.3℃。hrmsesi[m+h]+:calc.480.1957,found480.1964;1hnmr(400mhz,cdcl3):δ11.21(1h),8.71(1h),7.95-8.05(3h),7.90(1h),7.77(1h),7.40-7.70(3h),7.06(1h),4.03(2h),1.81(2h),1.54(2h),1.48(9h),1.00(3h)。验证例1:uv吸收光谱比较将实施例1、2、3、8及对照例1至2的紫外光吸收剂,配制成浓度20ppm的甲醇∶四氢呋喃=90∶10溶液,使用紫外/可见分光光谱仪(uv-2600;shimadzuinstrumentsco,ltd.)进行吸收度的量测,量测结果如图1、图2及表1所示。表1如图1的溶液中的uv吸收光谱所示,实施例1、2化合物和对照例1化合物相比,在吸收峰位置可以发现有明显红位移现象;此外,极限吸收波长位置也可以观察到实施例1、2化合物较对照例1、2化合物有较长波长的吸收位置,可以延伸到420nm之后。同时,与对照例2的苯环上5号位置不含氧原子取代基的化合物相比,实施例2的苯环上5号位置含有氧原子取代基的化合物,较长波长吸收峰位置(>350nm)有红位移现象,如表1所示。此结果表示,在苯并环上5号位置的含硫原子取代基与苯环上5号位置含有氧原子取代基,两者共存的情形下,可提供化合物优秀的红位移性质。此外,如图2所示,实施例2的化合物为苯环上5号位置的氧原子连接由羟基取代的烷基,实施例3的化合物为苯环上5号位置的氧原子连接由丙烯酸酯取代的烷基,两者在吸收峰位置仅有微小差异。实施例2的化合物为苯环上5号位置的氧原子连接由羟基取代的烷基,实施例8的化合物为苯环上5号位置的氧原子连接苄基,两者在吸收峰位置也几乎无改变。此结果显示,苯环上5号位置的氧原子所连接的不同反应性官能基团对uv光谱吸收峰位置的影响相当有限,原因是反应性官能基团并未直接键结于分子共轭结构上。光谱的主要差异为吸收强度的变化,来源为样品分子量间的差异与溶剂化作用间的差异影响。验证例2:穿透率测试将0.05克的实施例2或对照例1化合物与4克的pu主剂(型号a-7121)及2克乙酸乙酯混合均匀,再加入2克硬化剂(型号bayern75)混合均匀。后将混合完毕的pu胶以常规涂布机与涂布棒涂布在厚度为125μm的pet膜上,涂布后获得相应厚度为100μm湿膜,以80℃干燥30分钟,再进行穿透率量测。在此,pu薄膜中的穿透率已扣除基材pet膜的吸收。如图3的结果显示,实施例2化合物有明显的红位移现象,在400-450nm区间仍有一定的吸光能力。从图3的结果也可以看出对照例1化合物吸收极限位置位于约为400nm处,而实施例2化合物吸收极限位置则位于450nm之后。上述结果显示,实施例2化合物相较对照例1化合物具有明显红位移性质,也与溶液中的uv吸收结果相符合。验证例3:萃取测试将0.05克实施例2化合物或对照例1、3、4化合物加入6克pu主剂(型号a-7121)与3克溶剂混合均匀,再加入3克硬化剂(型号bayern75),均匀搅拌后置于室温4小时,后以80℃烘箱干燥16小时。加入30克的溶剂以超音波震荡萃取2小时后量测萃取率。其中,对照例3化合物使用重量比1比2的乙酸乙酯/甲苯混合溶剂,其余化合物使用乙酸乙酯为溶剂。结果如下表2所示。表2化合物分子量萃取率实施例2449.50%*对照例1451.699%对照例3658.973%对照例4479.694%*:0%表示未检出。如表2结果所示,反应型的实施例2化合物相较非反应型的对照例1化合物,虽然两者分子量相近,但抗萃取特性有显著差异。实施例2化合物在萃取实验中并未检出,而有明显的抗萃取特性;但对照例1化合物则是几乎全部被萃取出。另与分子量较大的对照例3化合物相比,实施例2化合物也显露优异的抗萃取性能,而对照例3化合物则是7成以上被萃取出。与有红位移特性的对照例4化合物相较,实施例2化合物也显示了较佳的抗萃取特性。验证例4:萃取测试将0.03克实施例3化合物或对照例1化合物加入0.9克1,6-己二醇二丙烯酸酯(1,6-hexanedioldiacrylate,hdda)、1.0克丙烯酸四氢呋喃酯(tetrahydrofurfurylacrylate,thfa)与0.07克1,1’-偶氮双(环己烷甲腈)(1,1’-azobis(cyclohexanecarbonitrile),abcn)的混合物中搅拌均匀,而后以80℃烘箱干燥24小时。加入10克的乙酸乙酯以超音波震荡萃取2小时后量测萃取率。结果如下表3所示。表3化合物分子量萃取率实施例3585.70.5%对照例1451.693.2%同样的,如表3结果所示,反应型的实施例3化合物相较非反应型的对照例1化合物,两者抗萃取特性有显著差异。实施例3化合物在萃取实验中仅有少量被萃取出,而有明显的抗萃取特性;但对照例1化合物则是大部分被萃取出。综上所述,本发明所提供的新颖化合物,其吸收光谱具有红位移的效果。当将本发明所提供的新颖化合物涂布于一对波长大于380nm的电磁辐射敏感的基材上时而形成一涂层时,该涂层可有效吸收大于380nm波长的光。因此,本发明所提供的新颖化合物可形成用于形成抗蓝光或抗紫外光的涂层,而提供一具有抗蓝光或抗紫外光效果的产品,例如,保护膜、镜片、隐形眼镜、人工水晶体等。此外,本发明所提供的新颖化合物,还具有抗萃取特性。当将本发明所提供的新颖化合物与高分子单体或寡聚物进行共聚生成含有紫外线吸收剂的高分子材料,高分子材料中的紫外线吸收剂因有良好的抗萃取特性,而可使高分子材料的应用领域更加广泛。上述实施例仅为了方便说明而举例而已,本发明所主张的权利范围自应以权利要求书所述为准,而非仅限于上述实施例。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1