基于捕获测序技术的肿瘤突变负荷检测装置及方法与流程
本发明涉及生物医学
技术领域:
,尤其涉及一种肿瘤突变负荷检测装置及方法。
背景技术:
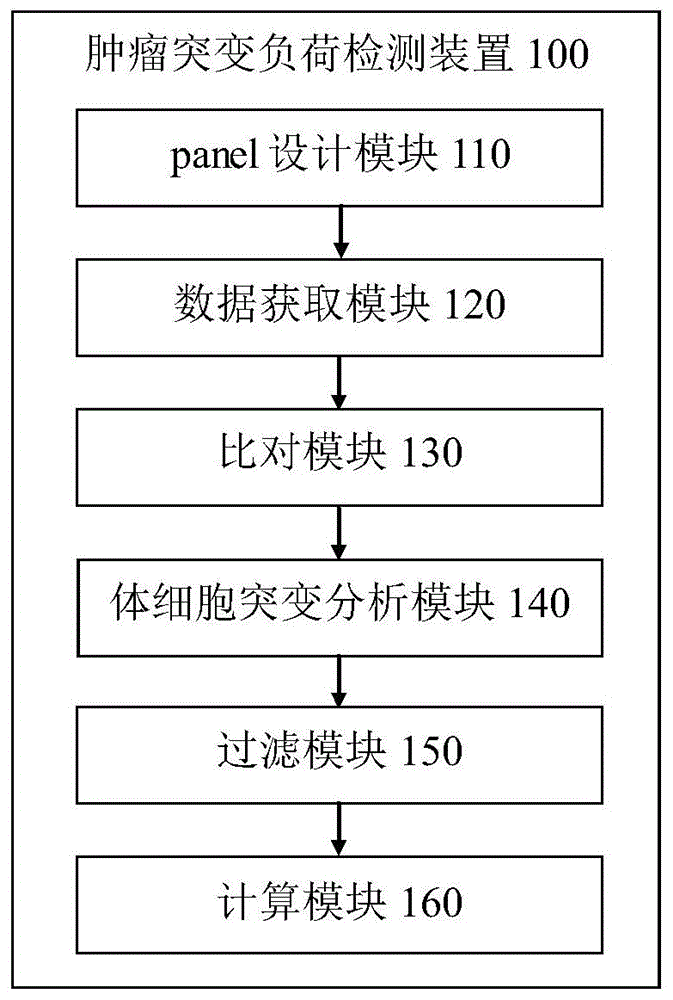
:肿瘤突变负荷,英文全称tumormutationburden(tmb)或tumormutationload(tml),是一种可定量的生物标志物,用来反映肿瘤细胞中含有的突变数目,通常用肿瘤细胞基因组编码区的每百万碱基突变数来衡量。现阶段对tmb检测主要依赖于ngs技术,金标准是通过wes测序(全外显子组测序技术)对≥30mb的cds区域(蛋白质编码区,外显子)序列中的突变数量进行统计分析与计算。然而全外显子检测存在价格昂贵、检测深度低、对于低覆盖的位点可能漏检等技术问题,因此研究者们积极探索基于捕获测序(panel)的方法对tmb进行检测,以有效降低测序成本,但是基于panel方法检测tmb时准确性和可靠性都存在较大挑战。目前,依然存在panel与全外显子测序一致性不够高、无对照样本检测结果时不准确、仅能单独检测肿瘤组织或者肿瘤患者血浆肿瘤突变负荷、对不同的测序深度的样本针对性差、对不同肿瘤占比的样本针对性差等缺点。技术实现要素:针对上述问题,本发明提供了一种基于捕获测序技术的肿瘤突变负荷检测装置及方法,有效解决现有检测技术中存在的panel与全外显子测序一致性不够高、仅能单独检测肿瘤组织或者肿瘤患者血浆肿瘤突变负荷等缺点。本发明提供的技术方案如下:一种基于捕获测序技术的肿瘤突变负荷检测装置,包括:panel设计模块,用于在基因组中均匀增加人群snp位点,并筛选与全外显子测序(wes)一致性最高的基因区域;数据获取模块,用于获取目标对象的组织和血浆样本,并基于所述panel设计模块筛选得到的基因区域获取所述组织和血浆样本的测序数据;比对模块,用于将所述数据获取模块获取的测序数据与参考基因组进行比对,获取变异数据结果;体细胞突变分析模块,用于对所述比对模块获取的变异数据结果进行体细胞分析得到体细胞突变结果;过滤模块,用于去除体细胞突变分析模块分析得到的体细胞突变结果中的非真实突变位点得到真实突变位点;及计算模块,用于根据所述过滤模块得到的体细胞真实突变位点数量计算肿瘤突变负荷tmb。本发明还提供了一种基于捕获测序技术的肿瘤突变负荷检测方法,包括:在基因组中均匀增加人群snp位点,并筛选与全外显子测序一致性最高的基因区域;获取目标对象的组织和血浆样本,并基于筛选得到的基因区域获取所述组织和血浆样本的测序数据;将所述测序数据与参考基因组进行比对,获取变异数据结果;对所述变异数据结果进行体细胞分析得到体细胞突变结果;去除所述体细胞突变结果中的非真实突变位点得到真实突变位点;根据所述体细胞真实突变位点数量计算肿瘤突变负荷tmb。本发明还提供了一种终端设备,包括存储器、处理器以及存储在所述存储器中并可在所述处理器上运行的计算机程序,所述处理器运行所述计算机程序时实现上述基于捕获测序技术的肿瘤突变负荷检测方法的步骤。本发明还提供了一种计算机可读存储介质,所述计算机可读存储介质存储有计算机程序,所述计算机程序被处理器执行时实现上述基于捕获测序技术的肿瘤突变负荷检测方法的步骤。本发明提供的基于捕获测序技术的肿瘤突变负荷检测装置及方法,在充分提高设计panel与wes的tmb一致性的前提下,提高panel设计的针对性、准确性和可靠性,尤其提高对于无对照样本结果的检测准确性,且能够同时检测肿瘤组织和肿瘤患者血浆的肿瘤突变负荷。具体,在panel设计方面通过均匀增加足够的人群snp位点来更准确地扣除胚系突变并使用基于机器学习新区间的筛选方法挑选与wes一致性最高的基因区域组合;另外,针对不同的深度测序、不同的样本类型和不同的肿瘤占比区间构建特异性基线,以此提高检测的适应性和准确性;再有,通过扣除序列特异性错误、测序或者实验背景噪音、突变黑名单和pon位点等,得到可信度高的体细胞变异信息;最后,能够对组织样本和血浆样本的测序数据同时进行检测处理,实现了对目标对象的组织和血浆样本的肿瘤突变负荷的同时检测且准确性较高。附图说明下面将以明确易懂的方式,结合附图说明优选实施方式,对上述特性、技术特征、优点及其实现方式予以进一步说明。图1为本发明中基于捕获测序技术的肿瘤突变负荷检测装置结构示意图;图2为本发明中基于捕获测序技术的肿瘤突变负荷检测方法流程示意图;图3为本发明一实例中肿瘤突变负荷检测流程图;图4为本发明一实例中全外显子和panel捕获得到的肿瘤突变负荷一致性结果示意图;图5为本发明中终端设备结构示意图。附图标记:100-肿瘤突变负荷检测装置,110-panel设计模块,120-数据获取模块,130-比对模块,140-体细胞突变分析模块,150-过滤模块,160-计算模块。具体实施方式为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对照附图说明本发明的具体实施方式。显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图,并获得其他的实施方式。本发明的第一实施例,如图1所示,一种基于捕获测序技术的肿瘤突变负荷检测装置100,包括:panel设计模块110,用于在基因组中均匀增加人群snp位点,并筛选与全外显子测序一致性最高的基因区域;数据获取模块120,用于获取目标对象的组织和血浆样本,并基于panel设计模块110筛选得到的基因区域获取组织和血浆样本的测序数据;比对模块130,用于将数据获取模块120获取的测序数据与参考基因组进行比对,获取变异数据结果;体细胞突变分析模块140,用于对比对模块130获取的变异数据结果进行体细胞分析得到体细胞突变结果;过滤模块150,用于去除体细胞突变分析模块140分析得到的体细胞突变结果中的非真实突变位点得到真实突变位点;及计算模块160,用于根据过滤模块150得到的体细胞真实突变位点数量计算肿瘤突变负荷tmb。在本实施例中,panel设计模块110用于筛选与wes一致性最高的基因区域组成panel,包括均匀位点设计单元和区间筛选单元,其中,均匀位点设计单元用于根据第一预设规则对基因组设计探针的区域进行筛选后均匀增加由第二预设规则筛选后得到的人群snp位点,以准确扣除胚系突变。区间筛选单元用于根据机器学习外显子exon的方法筛选得到与全外显子测序一致性最高的基因区域。由于现实情况,很多时候不能得到患者的血细胞数据,而tmb只考虑体细胞突变,所以多数tmb方法是在没有胚系对照数据的情况下,因此,为了提高使用insilico的算法去除可能的胚系突变过程中的准确度,本实施例在panel设计阶段均匀增加足够的人群snp位点。具体来说,设计包括以下步骤:1.1对基因组设计探针的区域进行筛选,所筛选的条件包括:去掉基因组上的gap以及mappability质量低于40的区域;将基因组按照预设大小的窗口(如200bp、300bp等)和步长(如1bp、2bp等)分割后,去除gc含量高于60%及低于30%的区域;1.2去除包含预设数量(如3等)以上亚洲人群杂合率大于预设阈值(如0.5、0.6等)的位点相应的预设长度(如120bp)区域;1.3对于进行探针设计的区域中千人基因组数据库中的snp位点进行筛选,筛选的条件包括:i)亚洲人群的杂合率大于某一阈值(如0.5、0.6等)的snp位点;ii)满足哈温平衡的snp位点;iii)将snp位点左右延长足够大小(如固定大小为100bp,且尽量使snp位点处在区域中间位置)方便设计探针;iv)使用现有成熟工具(如bwa,blast等)将上述延长后的区域与人类参考基因组序列比对,并统计每个区域可比对到基因组位置的数量,将数量大于预设阈值(如10个等)的区域去除。更进一步来说,过滤杂合率和哈温平衡的步骤如下:1)下载千人基因组phase3的snp数据;2)使用现有成熟工具(如plink)计算每个人群多态性位点的eas人群(千人基因组数据库中的亚洲人群数据)的最小等位基因频率(maf),以及哈温平衡的pvalue;3)过滤得到哈温平衡的pvalue大于某一固定阈值(如0.05、0.06等)的位点;4)筛选eas人群中maf较高的人群多态性位点。为了设计与wes一致性最高的panel,区间筛选单元的筛选过程包括:2.1对任一癌肿,在tcga或其他公共数据库(或自产样本数据库)中下载对应癌肿的dna突变数据;2.2下载人类基因组参考序列(hg19)及相应的注释文件,并按照注释文件的位置信息,统计每个样本每个exon上发生突变的个数(去除cosmic等致病突变),并标准化exon长度;2.3计算每个样本wes上的tmb值(记为tmb_wes);2.4去除gc含量(如去除gc含量高于60%及低于30%的区域)和mappability等不能设计探针的exon;2.5使用机器学习的方法对全部的exon进行排序,并依次标记为exon(1)、exon(2)、exon(3)、…、exon(n),其中,n为纳入分析的exon个数。挑选tmb-high(如tmb>10个/mb最高的样本)和tmb-low(如tmb<5个/mb值低的样本)肿瘤样本来对exon做排序。排序方法具体为:每次随机抽取一定比例(如70%、80%等)的样本做特征筛选,并重复多次(如100次、150次等),统计每个exon被挑中次数times,并按统计的times从大到小排序。特征筛选可以使用随机森林、logistics回归向后逐步回归等方法并以aic检验准则检验。在使用随机森林方法时,当exon被挑中的times一致时,还可以按重要性从大到小进行排序。2.6根据重要性排序后,从最重要的exon(1)开始,依次增加下一标记的exon,并计算每次exon集合的tmb值,并与wes的tmb结果的一致性进行评估(当下载的为tcga数据,则将其与tcgawes的tmb结果的一致性进行评估),当达到某一一致性阈值,或者通过增加exon已经不能有效提高一致性时,或者设定的区间大小已经差不多是最大可接受区间大小时停止计算,将该区间作为与wes一致性最高的基因区域。具体步骤如下:i)令挑选的exon区间集合记为exonset,且在第i轮中,exon_set={exon(1),…,exon(i)};ii)计算样本中仅包含exonset区间的tmb值(记为tmb_select_i);iii)如果满足下列条件之一,停止循环:a)tmb_select_i和tmb_wes之间的相关性cor(i)大于给定阈值(如r^2>0.9);b)cor(i)与cor(i-1)之间的差小于给定阈值(如0.0001等);c)exon_set中包含的exon的总长度大于给定阈值(如10m等);iv)如果步骤iii)未停止循环,则令exon_set={exon(1),…,exon(i),exon(i+1)},并重复步骤i)-iv)直到步骤iii)中停止循环。应当注意的是,在步骤iii)中b)可选的判断方法包括,直接计算排序下全部exon个数组合的相关性,并以曲线图形展示,当视觉上可见的达到某一exon个数时,相关性达到收敛条件,则选择达到收敛时的exon个数组合作为与wes一致性最高的基因区域。数据获取模块120包括获取单元和质控单元,其中,获取单元用于获取目标对象的组织和血浆样本的原始数据;质控单元用于分别对组织和血浆样本的原始数据进行质控处理,得到测序数据。比对模块130包括第一比对单元和第二比对单元,其中,第一比对单元用于将测序数据与参考基因组进行比对,得到比对结果文件;第二比对单元,用于对比对结果文件进行去冗余及针对indel区域进行重新比对,得到变异数据结果。在一实例中,第一对比单元中使用bwa软件将满足数据测序质量和测序数据质量的测序数据与人类参考基因组hg19进行比对,并用samtools软件对bam进行排序,得到变异数据结果;第二对比单元中用gatk和picard工具进行去冗余及indel区域重比对。在另一实例中,肿瘤突变负荷检测装置100还包括特异性基线构建模块,用于针对不同的测序深度区间、样本类型和肿瘤占比区间分别构建不同的测序深度基线和肿瘤占比基线。考虑到不同的测序深度或者样本类型,在覆盖度上可能存在不同的偏性,且在germlinesnp位点上,baf-0.5的偏差可能都会有所不同,故本实施例中针对会用到的不同测序深度或者样本类型构建不同的基线,已达到更好的适应性和准确性。另外,考虑到不同的组织样本病理切片中不同肿瘤占比导致的检测频率差异问题,本实施例中针对会不同的肿瘤占比区间构建不同的频率基线,以更灵敏更准确地用于不同纯度组织样本的真实突变鉴定。在一实例中,将现有肿瘤样本再病理评估中的照肿瘤占比的不同划分为多个不同的梯度,分别为0%-10%,10%-20%,20%-30%,30%以上,进而针对不同的肿瘤占比区间分别设置基线,使得tmb算法适用于不同肿瘤占比的病理样本。基于此,在体细胞突变分析模块140中,当有对照分析的样本时,使用vardict或mutect2对比对模块130获取的变异数据结果进行体细胞分析得到体细胞突变结果。当没有对照分析的样本时,根据组织和血浆样本的测序深度与样本类型,选择相应的测序深度基线,基于insilico胚系扣除算法得到体细胞突变结果。具体,在insilico胚系扣除算法的步骤具体包括:3.1采用mutect2等第三方软件检测全部候选的小突变,包括体细胞(somatic)的单碱基突变(snv)和胚系的单碱基突变(snp);3.2采用rollingmedian、局部加权回归法等方法统计覆盖率coverage,并做gc校正;3.3用健康人/已知阴性ffpe样本,构建不同测序深度、样本类型情况下的coverage的基线分布baseline1;3.4用健康人/已知阴性ffpe样本,构建不同测序深度,样本类型情况下的杂合snp的baf的基线,具体使用gatk等软件检测每个样本在每个snp位点的基因型,并分别统计杂合snpbaf的分布baseline2_1(均值μ,标准差σ,去除μ明显偏离0.5或方差过大的杂合snp),纯和snpbaf的分布baseline2_2,及无突变baf的分布baseline2_3;3.5使用深度/样本类型相对应的baseline1,计算待测样本每个捕获区间的拷贝数的log-ratio;3.6使用循环二元分割(cbs)方法对上述每个区间的log-ratio做分割segmentation。为方便表述,假设得到l个分割区域segment,在实例中,可以是带权重的cbs,如以健康人群覆盖度标准差的倒数为权重;3.7在得到的每个分割区域segment上,使用其上的snp位点做更细化的分割segmentation:a)snp位点要满足过滤条件:待测样本的max{baseline2_3}+k*σ<baf<min{baseline2_2}–k*σ,k=0、1、2或3,且覆盖深度大于某一阈值(如100);b)根据式(1)将每个baf转化为z-mbaf;z-mbaf=abs(baf-μ)/σ(1)c)对z-mbaf用cbs方法得到新的分割区域segment,假设最终得到m个分割区域segment。3.8在purecn、ascat等方法的基础上,使用网格搜索的方法估算肿瘤纯度(purity,ρ)和倍性(polidy,ψ)的多组局部最优解,并计算不同组合下拷贝数和baf的后验概率。定义mbaf=min{abs(baf-μ)+μ,100},使用log-ratio(ri)和mbaf(bi)来估算,其中,i表示第i个segment,变量ri和bi的期望如式(2)和式(3):其中,ci为拷贝数,且ci=na,i+nb,i,na,i和nb,i两个等位基因(allele)的拷贝数。3.9根据全部分割区域segment,使用最小二乘法求解ρ和ψ,同时估算基于拷贝数的信息(公式2)和基于snp的信息(公式3),并给予不同的权重。3.10根据估算的多个局部最优purity和ploidy组合和segment划分,使用purecn等软件判断每个候选snvsomatic的状态。基本原理是,根据beta分布先计算每个候选snv的log-likelihood,具此计算每个purity和ploidy组合的得分,并排序,通常最终选择得分最高的purity和ploidy组合,或根据经验的选择第二/第三排序的组合。体细胞突变分析模块140分析得到体细胞突变结果之后,过滤模块150随即针对体细胞突变分析模块140分析得到的体细胞突变结果的注释结果进行过滤去除其中的非真实突变位点得到数量为mn的真实突变位点。具体,过滤规则包括:根据样本类型去除insilico胚系突变;过滤注释频率小于5%且在人群数据库中出现频率大于0.2%的位点;过滤已知的肿瘤驱动基因突变;过滤突变位点表现为人群频率高的非胚系位点;和/或根据预先构建的ffpe样本特征sse的噪音基线过滤repeat区间或是同源区间比对产生的假阳性位点;和/或过滤频率小于pon位点均值加5倍标准差的pon位点;和/或过滤预设黑名单位点,人群出现频率大于30%或者在ffpe样本、血浆样本和血细胞样本中的两个组织类型里面人群频率大于20%的位点;和/或根据测序深度基线筛选符合深度要求的突变,根据肿瘤占比基线得到符合肿瘤占比的突变。在一实例中,使用mutect2对变异数据结果进行体细胞分析,得到vcf文件结果(体细胞突变结果)后,使用annovar软件进行注释,得到数据库注释结果;进而过滤模块150针对注释位点进行过滤。具体,这一过程中,为了严格控制纳入计算的突变位点,同时考虑了测序或者实验背景噪音、序列特异性错误产生的突变,pon以及位点黑名单进行假阳性过滤,最终得到高可信度的体细胞变异信息。主要分为以下几个步骤:4.1背景噪音根据一定数量(如30)正常人突变位点的频率(大于等于0.1%)分布,选取单侧95%的置信区间作为背景噪音的阈值,样本位点突变频率大于等于均值加3倍标准差(mean+3sd)保留。4.2sse(序列特异性错误)导致的假阳性突变过滤突变位点表现为人群频率高的非胚系位点、repeat区间或者是同源区间比对产生的假阳性位点,通过建立ffpe样本特征sse的噪音基线,严格过滤sse。4.3panelofnormals(pon)用相同的实验以及分析流程对一定数量(如30)正常人血细胞和血浆样本,分别进行突变位点的出现频率统计,有两个及以上正常人出现的位点作为pon位点,对于在pon范围的突变,实际检测样本频率大于等于pon位点均值加5倍标准差则保留,否则将被过滤掉。4.4黑名单取内部数据库一定数量(如1000)例ffpe样本、血浆样本以及血细胞样本构建突变黑名单,统计各个突变在人群中的出现频率,选取人群出现频率大于30%或者在任何两个组织类型里面人群频率都大于20%的位点作为黑名单位点,黑名单位点将被直接过滤掉。以此计算模块160根据过滤模块150得到的体细胞真实突变位点数量计算肿瘤突变负荷tmb,如式(4):tmb=mn/tn*1000000(4)其中,tn表示所有变异数据中突变位点的数量。在上述实施例中,克服了目前tmb检测方法存在的针对性较低、一致性不高、可靠性不高、对无对照样本结果检测结果不准确、仅能单独检测肿瘤组织或者肿瘤患者血浆肿瘤突变负荷等缺陷,其在充分提高设计panel与wes的tmb一致性的前提下,全面提高各个环节的准确性,尤其提高panel设计的针对性、准确性和可靠性;提高对于无对照样本结果的检测准确性;提高不同深度、不同纯度、不同肿瘤占比的特殊组织或血浆样本的检测准确性,为tmb的计算提供了一种针对性更强、敏感度更高、准确度更高的检测装置。本发明的另一实施例中,如图2所示,一种基于捕获测序技术的肿瘤突变负荷检测方法,可应用于上述肿瘤突变负荷检测装置,该肿瘤突变负荷检测方法包括:s10在基因组中均匀增加人群snp位点,并筛选与全外显子测序一致性最高的基因区域;s20获取目标对象的组织和血浆样本,并基于筛选得到的基因区域获取组织和血浆样本的测序数据;s30将测序数据与参考基因组进行比对,获取变异数据结果;s40对变异数据结果进行体细胞分析得到体细胞突变结果;s50去除体细胞突变结果中的非真实突变位点得到真实突变位点;s60根据体细胞真实突变位点数量计算肿瘤突变负荷tmb。在本实施例中,由于现实情况,很多时候不能得到患者的血细胞数据,而tmb只考虑体细胞突变,所以多数tmb方法是在没有胚系对照数据的情况下,因此,为了提高使用insilico的算法去除可能的胚系突变过程中的准确度,本实施例在panel设计阶段均匀增加足够的人群snp位点。具体来说,设计包括以下步骤:1.1对基因组设计探针的区域进行筛选,所筛选的条件包括:去掉基因组上的gap以及mappability质量低于40的区域;将基因组按照预设大小的窗口(如200bp、300bp等)和步长(如1bp、2bp等)分割后,去除gc含量高于60%及低于30%的区域;1.2去除包含预设数量(如3等)以上亚洲人群杂合率大于预设阈值(如0.5、0.6等)的位点相应的预设长度(如120bp)区域;1.3对于进行探针设计的区域中千人基因组数据库中的snp位点进行筛选,筛选的条件包括:i)亚洲人群的杂合率大于某一阈值(如0.5、0.6等)的snp位点;ii)满足哈温平衡的snp位点;iii)将snp位点左右延长足够大小(如固定大小为100bp,且尽量使snp位点处在区域中间位置)方便设计探针;iv)使用现有成熟工具(如bwa,blast等)将上述延长后的区域与人类参考基因组序列比对,并统计每个区域可比对到基因组位置的数量,将数量大于预设阈值(如10个等)的区域去除。为了设计与wes一致性最高的panel,区间筛选单元的筛选过程包括:2.1对任一癌肿,在tcga或其他公共数据库(或自产样本数据库)中下载对应癌肿的dna突变数据;2.2下载人类基因组参考序列(hg19)及相应的注释文件,并按照注释文件的位置信息,统计每个样本每个exon上发生突变的个数(去除cosmic等致病突变),并标准化exon长度;2.3计算每个样本wes上的tmb值(记为tmb_wes);2.4去除gc含量(如去除gc含量高于60%及低于30%的区域)和mappability等不能设计探针的exon;2.5使用机器学习的方法对全部的exon进行排序,并依次标记为exon(1)、exon(2)、exon(3)、…、exon(n),其中,n为纳入分析的exon个数。挑选tmb-high(如tmb>10个/mb最高的样本)和tmb-low(如tmb<5个/mb值低的样本)肿瘤样本来对exon做排序。排序方法具体为:每次随机抽取一定比例(如70%、80%等)的样本做特征筛选,并重复多次(如100次、150次等),统计每个exon被挑中次数times,并按统计的times从大到小排序。特征筛选可以使用随机森林、logistics回归向后逐步回归等方法并以aic检验准则检验。在使用随机森林方法时,当exon被挑中的times一致时,还可以按重要性从大到小进行排序。2.6根据重要性排序后,从最重要的exon(1)开始,依次增加下一标记的exon,并计算每次exon集合的tmb值,并与wes的tmb结果的一致性进行评估(当下载的为tcga数据,则将其与tcgawes的tmb结果的一致性进行评估),当达到某一一致性阈值,或者通过增加exon已经不能有效提高一致性时,或者设定的区间大小已经差不多是最大可接受区间大小时停止计算,将该区间作为与wes一致性最高的基因区域。具体步骤如下:i)令挑选的exon区间集合记为exonset,且在第i轮中,exon_set={exon(1),…,exon(i)};ii)计算样本中仅包含exonset区间的tmb值(记为tmb_select_i);iii)如果满足下列条件之一,停止循环:a)tmb_select_i和tmb_wes之间的相关性cor(i)大于给定阈值(如r^2>0.9);b)cor(i)与cor(i-1)之间的差小于给定阈值(如0.0001等);c)exon_set中包含的exon的总长度大于给定阈值(如10m等);iv)如果步骤iii)未停止循环,则令exon_set={exon(1),…,exon(i),exon(i+1)},并重复步骤i)-iv)直到步骤iii)中停止循环。应当注意的是,在步骤iii)中b)可选的判断方法包括,直接计算排序下全部exon个数组合的相关性,并以曲线图形展示,当视觉上可见的达到某一exon个数时,相关性达到收敛条件,则选择达到收敛时的exon个数组合作为与wes一致性最高的基因区域。在步骤s20中,获取目标对象的组织和血浆样本的原始数据的之后,分别对其进行质控处理,得到测序数据。在步骤s30中,首先将测序数据与参考基因组进行比对,得到比对结果文件;之后对比对结果文件进行去冗余及针对indel区域进行重新比对,得到变异数据结果。在一实例中,使用bwa软件将满足数据测序质量和测序数据质量的测序数据与人类参考基因组hg19进行比对,并用samtools软件对bam进行排序,得到变异数据结果;使用gatk和picard工具进行去冗余及indel区域重比对。在另一实例中,基于捕获测序技术的肿瘤突变负荷检测方法中还包括针对不同的测序深度区间、样本类型和肿瘤占比区间分别构建不同的测序深度基线和肿瘤占比基线的步骤。具体,考虑到不同的测序深度或者样本类型,在覆盖度上可能存在不同的偏性,且在germlinesnp位点上,baf-0.5的偏差可能都会有所不同,故本实施例中针对会用到的不同测序深度或者样本类型构建不同的基线,已达到更好的适应性和准确性。另外,考虑到不同的组织样本病理切片中不同肿瘤占比导致的检测频率差异问题,本实施例中针对会不同的肿瘤占比区间构建不同的频率基线,以更灵敏更准确地用于不同纯度组织样本的真实突变鉴定。在一实例中,将现有肿瘤样本再病理评估中的照肿瘤占比的不同划分为多个不同的梯度,分别为0%-10%,10%-20%,20%-30%,30%以上,进而针对不同的肿瘤占比区间分别设置基线,使得tmb算法适用于不同肿瘤占比的病理样本。基于此,在步骤s40中,当有对照分析的样本时,使用vardict或mutect2对变异数据结果进行体细胞分析得到体细胞突变结果。当没有对照分析的样本时,根据组织和血浆样本的测序深度与样本类型,选择相应的测序深度基线,基于insilico胚系扣除算法得到体细胞突变结果。具体,在insilico胚系扣除算法的步骤具体包括:3.1采用mutect2等第三方软件检测全部候选的小突变,包括体细胞(somatic)的单碱基突变(snv)和胚系的单碱基突变(snp);3.2采用rollingmedian、局部加权回归法等方法统计覆盖率coverage,并做gc校正;3.3用健康人/已知阴性ffpe样本,构建不同测序深度、样本类型情况下的coverage的基线分布baseline1;3.4用健康人/已知阴性ffpe样本,构建不同测序深度,样本类型情况下的杂合snp的baf的基线,具体使用gatk等软件检测每个样本在每个snp位点的基因型,并分别统计杂合snpbaf的分布baseline2_1(均值μ,标准差σ,去除μ明显偏离0.5或方差过大的杂合snp),纯和snpbaf的分布baseline2_2,及无突变baf的分布baseline2_3;3.5使用深度/样本类型相对应的baseline1,计算待测样本每个捕获区间的拷贝数的log-ratio;3.6使用循环二元分割(cbs)方法对上述每个区间的log-ratio做分割segmentation。为方便表述,假设得到l个分割区域segment,在实例中,可以是带权重的cbs,如以健康人群覆盖度标准差的倒数为权重;3.7在得到的每个分割区域segment上,使用其上的snp位点做更细化的分割segmentation:a)snp位点要满足过滤条件:待测样本的max{baseline2_3}+k*σ<baf<min{baseline2_2}–k*σ,k=0、1、2或3,且覆盖深度大于某一阈值(如100);b)根据式(1)将每个baf转化为z-mbaf;c)对z-mbaf用cbs方法得到新的分割区域segment,假设最终得到m个分割区域segment。3.8在purecn、ascat等方法的基础上,使用网格搜索的方法估算肿瘤纯度(purity,ρ)和倍性(polidy,ψ)的多组局部最优解,并计算不同组合下拷贝数和baf的后验概率。定义mbaf=min{abs(baf-μ)+μ,100},使用log-ratio(ri)和mbaf(bi)来估算,其中,i表示第i个segment,变量ri和bi的期望如式(2)和式(3)。3.9根据全部分割区域segment,使用最小二乘法求解ρ和ψ,同时估算基于拷贝数的信息(公式2)和基于snp的信息(公式3),并给予不同的权重。3.10根据估算的多个局部最优purity和ploidy组合和segment划分,使用purecn等软件判断每个候选snvsomatic的状态。基本原理是,根据beta分布先计算每个候选snv的log-likelihood,具此计算每个purity和ploidy组合的得分,并排序,通常最终选择得分最高的purity和ploidy组合,或根据经验的选择第二/第三排序的组合。得到体细胞突变结果之后,随即步骤s50中针对得到的体细胞突变结果的注释结果进行过滤去除其中的非真实突变位点得到数量为mn的真实突变位点。具体,过滤规则包括:根据样本类型去除insilico胚系突变;过滤注释频率小于5%且在人群数据库中出现频率大于0.2%的位点;过滤已知的肿瘤驱动基因突变;过滤突变位点表现为人群频率高的非胚系位点;和/或根据预先构建的ffpe样本特征sse的噪音基线过滤repeat区间或是同源区间比对产生的假阳性位点;和/或过滤频率小于pon位点均值加5倍标准差的pon位点;和/或过滤预设黑名单位点,人群出现频率大于30%或者在ffpe样本、血浆样本和血细胞样本中的两个组织类型里面人群频率大于20%的位点;和/或根据测序深度基线筛选符合深度要求的突变,根据肿瘤占比基线得到符合肿瘤占比的突变。在一实例中,使用mutect2对变异数据结果进行体细胞分析,得到vcf文件结果(体细胞突变结果)后,使用annovar软件进行注释,得到数据库注释结果;进而在步骤s50中,针对注释位点进行过滤。以此在步骤s60中根据过滤模块得到的体细胞真实突变位点数量计算肿瘤突变负荷tmb,如式(4)。在一实例中:一、测序文库构建基于ngs测序方法,组织样本(ffpe)、血浆样本和血细胞样本(bc)进行文库构建,建库步骤如下(其中血细胞样本不需要打断处理):1.样本打断:将聚四氟乙烯线用紫外灭菌后的医用剪刀,剪至1cm左右的长度,并且保证打断棒的长度均一性良好,置于干净容器中,紫外灭菌3-4小时。灭菌完成后,将1cm的聚四氟乙烯线,用灭菌后的镊子装进96孔板内。每个孔装入2根打断棒,完成后再将96孔板紫外灭菌3-4小时。按照qubit定量结果取300ngffpe/bcdna样本,使用te稀释到50μl,转移到96孔板中,将锡箔纸膜放在96孔板上,四边对齐,使用热封膜仪180℃5s封膜2次,使用微孔板离心机离心。选择预先设定的程序peakpower:450;dutyfactor:30;cycles/burst:200;treatmenttime:40s,3cycles,点击“startposition”。在run界面点“run”按钮,运行程序。在该程序运行完成后,取出样品板,使用微孔板离心机离心,再将样品板放到样品架上,选择程序peakpower:450;dutyfactor:30;cycles/burst:200;treatmenttime:40s,4cycles。在run界面点“run”按钮,运行程序。在该程序运行完成后,取出样品板,使用微孔板离心机离心。打断后取1μl进行质检。2.文库制备步骤:末端修复并在3’末端加a尾:按照下表1配制er﹠atmix。表1:er﹠atmix配制试剂体积endrepair&a-tailingbuffer7μlendrepair&a-tailingenzymemix3μl总体积10μl取10μler﹠atmix加入dna样本中(冰上操作),震荡混匀,短暂离心。注意er﹠atmix与dna涡旋混匀立即进行pcr反应。反应体系置于pcr仪上,按下表进行pcr反应。这里pcr仪热盖温度设为85℃。若该操作结束后立即进行下表2所示步骤实验,应将终止温度设为20℃。表2:末端修复和加a尾实验条件连接接头:adapter准备:idtudiadapte2.5μl,加2.5ul水稀释到5μl。配制ligationmix(冰上操作):根据文库个数,按照下表3配制ligationmix,震荡混匀。表3:ligationmix配制试剂体积超纯水5μlligationbuffer30μldnaligase10μl总体积45μl上一步pcr结束后,取出样本。短暂离心,转入稀释好的adapter溶液中。然后加入45μlligationmix,震荡混匀,短暂离心。置于pcr仪上,20℃孵育30min,20℃保存,热盖温度为50℃。连接后纯化:上一步pcr结束后取出样本,短暂离心,加入88μl磁珠。震荡混匀(震荡时按紧管盖),室温孵育15min,使dna与磁珠充分结合。短暂离心,离心管置于磁力架上待液体澄清(不要吸到磁珠),弃去上清。加入200μl80%乙醇孵育30sec后弃去。重复一次200μl80%乙醇(现用现配)清洗步骤。用10μl枪头吸尽离心管底部的残留乙醇,室温干燥3-5min至乙醇完全挥发(正面看不在反光,背面看已经干燥)。注意:磁珠过分干燥dna产量会减少。从磁力架取下离心管,加入22μl超纯水,震荡混匀(震荡时按紧管盖)。室温孵育5min。短暂离心,离心管置于磁力架上待液体澄清。取1μldna文库用于浓度检测,剩余的20μl清液转移至新的pcr管进行下一步扩增试验。文库扩增:按照下表4配制pcrmix(冰上操作),震荡混匀。短暂离心,将pcrmix分装至0.2mlpcr管中,置于4℃冰箱保存。表4:pcrmix配制试剂体积hifihotstartreadymix(2×)25μllibraryamplificationprimermix(10×)5μl总体积30μl将上一步的文库转入已分装的pcrmix,震荡混匀。短暂离心,置于pcr仪上,按下表5进行pcr反应。表5:pcr反应反应条件dna的获得(1xbeads回收):pcr结束后,取出样本。短暂离心,加入50μlbeckmanagencourtampurexp磁珠。震荡混匀(震荡时按紧管盖),室温孵育15min,使dna与磁珠充分结合。短暂离心,离心管置于磁力架上待液体澄清(不要吸到磁珠),弃去上清。加入200μl80%乙醇(现用现配)孵育30sec后弃去。重复一次200μl80%乙醇清洗步骤。用10μl枪头吸尽离心管底部的残留乙醇,室温干燥3-5min至乙醇完全挥发(正面看不在反光,背面看已经干燥)。注意:磁珠过分干燥dna产量会减少。从磁力架取下离心管,加入40μl超纯水,振荡混匀。室温孵育5min洗脱dna。短暂离心,离心管置于磁力架上待液体澄清,将文库转移至新的离心管中。保存于-20℃。3.文库质检:取1μldna文库用于浓度检测。基于ngs测序方法,ffpe、血浆和bc样本的捕获如下:选取370个基因进行全外捕获,覆盖外显子区域1684573bp,具体基因列表见表10。4.混合文库:取总量1μg的等量文库于1.5ml离心管中,根据每个文库的浓度和capture文库个数计算每个文库加入的体积。文库加入的体积是:(1000ng/capture文库个数/文库浓度)μl。加入universalblockingoligos向上述体系中加入2.5μluniversalblockingoligos。加入5μlcothumandna,震荡混匀,短暂离心。用封口膜封住ep管,放入真空离心浓缩仪中蒸干(60℃,约20min-1hr)。注意随时查看是否已蒸干。dna变性:样本完全蒸干后,每个capture中加入7.5μl2×hybridizationbuffer(vial5)和3μlhybridizationcomponenta(vial6),震荡混匀,短暂离心。置于95℃加热模块变性10min。5.文库与探针杂交取出探针短暂离心后置于47℃pcr仪中,迅速将变性的dna从95℃转移至含有探针的pcr管中,震荡混匀,短暂离心。置于pcr仪中,47℃杂交,杂交时间应不少于16hr。配制washbuffer工作液:一个capture所需缓冲液的配制方法如下表6,根据capture的个数按下表6配制缓冲液。表6:缓冲液配制试剂试剂/μl水/μl1×工作液体积/μl10×stringentwashbuffer(vial4)4036040010×washbufferⅰ(vial1)3027030010×washbufferⅱ(vial2)2018020010×washbufferiii(vial3)201802002.5×beadwashbuffer(vial7)200300500分装需要孵育的试剂:分装400μl1×stringentwashbuffer(vial4)至八连排中;分装100μl1×washbufferi(vial1)至八连排中;分装20μlcapturebeads至八连排中。孵育capturebeads和washbuffer(vial4和vial1)工作液:capturebeads使用前须室温平衡30min。washbuffer(vial4和vial1)工作液使用前须47℃孵育2hr。6.杂交后纯化:每个capture分装100μl捕获磁珠,将100μl捕获磁珠置于磁力架上至液体澄清,弃去上清。加入200μl1×beadwashbuffer(vial7),震荡混匀。置于磁力架上至液体澄清,弃去上清。加入200μl1×beadwashbuffer(vial7),震荡混匀。置于磁力架上至液体澄清,弃去上清。加入100μl1×beadwashbuffer(vial7),震荡混匀。置于磁力架上至液体澄清,弃去上清。此时磁珠预处理完成,立即进行下一步试验。将捕获过夜的杂交液体转入清洗好的磁珠中,移液器吹打十次。置于pcr仪中47℃孵育45min(pcr热盖温度设为57℃),每隔15min震荡一次保证磁珠悬浮。清洗:孵育完成后,每管加入100μl47℃预热的1×washbufferi(vial1),震荡混匀。置于磁力架上至液体澄清,弃去上清。加入200μl47℃预热的1×stringentwashbuffer(vial4),移液器吹打十次混匀。47℃孵育5min,置于磁力架上至液体澄清,弃去上清。注意操作过程尽量避免温度低于47℃。加入200μl47℃预热的1×stringentwashbuffer(vial4),移液器吹打十次混匀。47℃孵育5min,置于磁力架上至液体澄清,弃去上清。注意操作过程尽量避免温度低于47℃。加入200μl室温放置的1×washbufferi(vial1),振荡2min,短暂离心,置于磁力架上至液体澄清,弃去上清。加入200μl室温放置的1×washbufferii(vial2),震荡1min,短暂离心,放置磁力架上至液体澄清,弃去上清。加入200μl室温放置的1×washbufferiii(vial3),震荡30sec,短暂离心,放置磁力架上至液体澄清,弃去上清。向离心管中加入20μl超纯水洗脱,震荡混匀,进行下一步扩增试验。7.post-lm-pcr:按照表7配制post-lm-pcrmix,震荡混匀。表7:post-lm-pcrmix配制试剂体积hifihotstartreadymix25μlpost-lm-pcroligos1&2,5μm5μl上一步洗脱的dna20μltotal50μl将上述样本转入pcr反应中,震荡混匀,短暂离心。置于pcr仪上,按下表8进行pcr反应:表8:pcr反应条件扩增后纯化:取出纯化磁珠(dnapurificationbeads),室温平衡30min备用。取90μl纯化磁珠于1.5ml离心管中,加入50μl扩增后的捕获dna文库,振荡混匀,室温孵育15min。置于磁力架上至液体澄清,弃去上清。加入200μl80%乙醇(现用现配)孵育30sec后弃去。重复一次200μl80%乙醇清洗步骤。用10μl枪头弃去离心管底部的残留乙醇,室温干燥至乙醇完全挥发(前面看磁珠不反光,背面看干燥)。注意:磁珠过分干燥dna产量会减少。从磁力架取下离心管,加入50μl超纯水,振荡混匀。室温孵育2min。短暂离心,置于磁力架上至液体澄清,将capture样本转入新的离心管中。8.质检:取1μlcapture样本用于qubit浓度检测。文库库检合格后上机,上机平台选择illumina平台的nexseq500测序仪,测序策略为pe75,每个样本数据量为10g。二、数据分析具体分析流程图见附图3:5.1判断数据质控、数据测序质量及测序总量是否满足,若是,得到cleandata。5.2将得到的cleandata用bwa比对到人参考基因组hg19,用samtools对bam文件进行排序;5.3将得到的bam文件用picard和gatk工具进行去冗余及indel区域重比对;5.4将得到的重比对后的bam文件使用mutect2/vardict分析体细胞突变,得到vcf文件;5.5将得到的vcf文件用annovar工具做注释,得到数据库注释结果;5.6将得到注释文件,过滤频率小于5%,在人群数据库中出现频率大于0.2%位点,过滤掉明确已知的肿瘤驱动基因突变,过滤突变位点表现为人群频率高的非胚系位点、repeat区间或者是同源区间比对产生的假阳性位点,通过建立的ffpe样本特征sse噪音基线过滤sse;过滤pon位点:对于在pon范围的突变,实际检测样本频率大于等于pon位点均值加5倍标准差则保留;过滤黑名单位点;考虑样本的肿瘤占比所处的范围,根据不同的样本类型扣除insilico胚系突变,并根据测序深度基线筛选符合深度要求的突变;5.7将上述过滤得到最终用来纳入计算的体细胞突变位点计数为mn;5.8将5.3得到的bam文件用samtools工具得到每个位点的覆盖深度;5.9统计5.8统计的文件突变总数计数为tn,将上述过滤得到最终用来纳入计算的体细胞突变位点计数为mn;5.10对肿瘤突变负荷进行计算tmb=mn/tn*1000000。按照上述方法对37例患者的组织样本,分别做了全外显子测序和panel捕获测序,分析患者的肿瘤突变负荷,并分析这37例患者全外显子和panel捕获得到的肿瘤突变负荷一致性结果,结果见附图4(横坐标为wes检测的tmb,纵坐标为panel捕获测序检测的tmb),从图中可以看出,该37例患者全外显子和panel捕获得到的肿瘤突变负荷的相关性r^2=0.965。瘤突变负荷结果详细见下表9。表9:37例患者全外显子和panel捕获检测到的肿瘤突变负荷结果从以上结果可以看出,本申请的肿瘤突变负荷的检测方法不仅能够同时检测组织和血浆样本,而且检测结果准确性较高。表10:370个基因列表所属领域的技术人员可以清楚地了解到,为了描述的方便和简洁,仅以上述各程序模块的划分进行举例说明,实际应用中,可以根据需要而将上述功能分配由不同的程序模块完成,即将装置的内部结构划分成不同的程序单元或模块,以完成以上描述的全部或者部分功能。实施例中的各程序模块可以集成在一个处理单元中,也可是各个单元单独物理存在,也可以两个或两个以上单元集成在一个处理单元中,上述集成的单元既可以采用硬件的形式实现,也可以采用软件程序单元的形式实现。另外,各程序模块的具体名称也只是为了便于相互区分,并不用于限制本申请的保护范围。图5是本发明一个实施例中提供的终端设备的结构示意图,如所示,该终端设备200包括:处理器220、存储器210以及存储在存储器210中并可在处理器220上运行的计算机程序211,例如:基于捕获测序技术的肿瘤突变负荷检测方法关联程序。处理器220执行计算机程序211时实现上述各个基于捕获测序技术的肿瘤突变负荷检测方法实施例中的步骤,或者,处理器220执行计算机程序211时实现上述基于捕获测序技术的肿瘤突变负荷检测装置实施例中各模块的功能。终端设备200可以为笔记本、掌上电脑、平板型计算机、手机等设备。终端设备200可包括,但不仅限于处理器220、存储器210。本领域技术人员可以理解,图5仅仅是终端设备200的示例,并不构成对终端设备200的限定,可以包括比图示更多或更少的部件,或者组合某些部件,或者不同的部件,例如:终端设备200还可以包括输入输出设备、显示设备、网络接入设备、总线等。处理器220可以是中央处理单元(centralprocessingunit,cpu),还可以是其他通用处理器、数字信号处理器(digitalsignalprocessor,dsp)、专用集成电路(applicationspecificintegratedcircuit,asic)、现场可编程门阵列(field-programmablegatearray,fpga)或者其他可编程逻辑器件、分立门或者晶体管逻辑器件、分立硬件组件等。通用处理器220可以是微处理器或者该处理器也可以是任何常规的处理器等。存储器210可以是终端设备200的内部存储单元,例如:终端设备200的硬盘或内存。存储器210也可以是终端设备200的外部存储设备,例如:终端设备200上配备的插接式硬盘,智能存储卡(smartmediacard,smc),安全数字(securedigital,sd)卡,闪存卡(flashcard)等。进一步地,存储器210还可以既包括终端设备200的内部存储单元也包括外部存储设备。存储器210用于存储计算机程序211以及终端设备200所需要的其他程序和数据。存储器210还可以用于暂时地存储已经输出或者将要输出的数据。在上述实施例中,对各个实施例的描述都各有侧重,某个实施例中没有详细描述或记载的部分,可以参见其他实施例的相关描述。本领域普通技术人员可以意识到,结合本文中所公开的实施例描述的各示例的单元及算法步骤,能够以电子硬件、或者计算机软件和电子硬件的结合来实现。这些功能究竟以硬件还是软件来执行,取决于技术方案的特定应用和设计约束条件。专业技术人员可以对每个特定的应用来使用不同方法来实现所描述的功能,但是这种实现不应认为超出本申请的范围。在本申请所提供的实施例中,应该理解到,所揭露的装置/终端设备和方法,可以通过其他的方式实现。例如,以上所描述的装置/终端设备实施例仅仅是示意性的,例如,模块或单元的划分,仅仅为一种逻辑功能划分,实际实现时可以有另外的划分方式,例如,多个单元或组件可以结合或者可以集成到另一个系统,或一些特征可以忽略,或不执行。另一点,所显示或讨论的相互之间的耦合或直接耦合或通讯连接可以是通过一些接口,装置或单元的间接耦合或通讯连接,可以是电性、机械或其他的形式。作为分离部件说明的单元可以是或者也可以不是物理上分开的,作为单元显示的部件可以是或者也可以不是物理单元,即可以位于一个地方,或者也可以分布到多个网络单元上。可以根据实际的需要选择其中的部分或者全部单元来实现本实施例方案的目的。另外,在本申请各个实施例中的各功能单元可能集成在一个处理单元中,也可以是各个单元单独物理存在,也可以两个或两个以上单元集成在一个单元中。上述集成的单元既可以采用硬件的形式实现,也可以采用软件功能单元的形式实现。集成的模块/单元如果以软件功能单元的形式实现并作为独立的产品销售或使用时,可以存储在一个计算机可读存储介质中。基于这样的理解,本发明实现上述实施例方法中的全部或部分流程,也可以通过计算机程序211发送指令给相关的硬件完成,的计算机程序211可存储于一计算机可读存储介质中,该计算机程序211在被处理器220执行时,可实现上述各个方法实施例的步骤。其中,计算机程序211包括:计算机程序代码,计算机程序代码可以为源代码形式、对象代码形式、可执行文件或某些中间形式等。计算机可读存储介质可以包括:能够携带计算机程序211代码的任何实体或装置、记录介质、u盘、移动硬盘、磁碟、光盘、计算机存储器、只读存储器(rom,read-onlymemory)、随机存取存储器(ram,randomaccessmemory)、电载波信号、电信信号以及软件分发介质等。需要说明的是,计算机可读存储介质包含的内容可以根据司法管辖区内立法和专利实践的要求进行适当的增减,例如:在某些司法管辖区,根据立法和专利实践,计算机可读介质不包括电载波信号和电信信号。应当说明的是,上述实施例均可根据需要自由组合。以上仅是本发明的优选实施方式,应当指出,对于本
技术领域:
的普通相关人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1