一种磁性共价有机框架材料及其制备方法和应用与流程
本发明涉及吸附材料技术领域,具体涉及一种磁性共价有机框架材料及其制备方法和应用。
背景技术:
农药主要用于防治植物病、虫、草害,在农业生产中的广泛使用,不可避免地会在食品及水体中残留,对人体健康造成严重威胁。因此,开发高效的吸附移除材料显得尤为重要。共价有机框架材料(covalentorganicframeworks简称cof)是一类通过共价键连接的有机多孔框架聚合物,具有结构高度有序、比表面积大、化学稳定性及热稳定性好等诸多优点,可以提高其作为有机靶标吸附移除剂的承载能力,因此cof在分离化学领域,尤其是在水体中污染物移除领域具备很好的应用价值和潜力。然而,现有的cof以苯环为主要功能基团,疏水性过强,导致其在去除水体中污染物时分散性较差,进而影响吸附速率与容量,而且由于cof本身的密度低,通常为粉末堆积形态,即便是通过离心分离也难以保证cof在吸附完目标物之后从吸附介质中高效快速分离和回收。
为了解决传统cof材料存在的上述问题,通常对cof材料进行修饰。例如,shutingzhuang等(magneticcoffortheadsorptiveremovalofdiclofenacandsulfamethazinefromaqueoussolution:adsorptionkinetics,isothermsstudyanddftcalculation,journalofhazardousmaterials385(2020)121596.)利用2,5-二甲氧基苯-1,4-二甲醛(dmta)与1,3,5-三(4-氨基苯基)苯(tapb)通过胺醛缩合制备了磁性cof材料,实现了其对水溶液中双氯芬酸和磺胺甲嘧啶的吸附移除,然而,其制备的磁性cof材料中fe3o4嵌入cof孔道内,其饱和磁化强度仅为5.2emu/g,磁性较低,导致固液分散体系分离速率低;而且,由于磁性cof材料的亲水性差,有机污染物的吸附平衡时间长,吸附速率低。
技术实现要素:
鉴于此,本发明的目的在于提供一种磁性共价有机框架材料及其制备方法和应用,本发明提供的磁性共价有机框架材料亲水性强、对农药有机污染物的吸附性强、吸附效率高且易高效回收。
为了实现上述发明目的,本发明提供以下技术方案:
本发明提供了一种磁性共价有机框架材料,具有核-壳结构,所述核-壳结构中的核为fe3o4@sio2-nh2纳米微球,壳的化学组成为cof;所述cof以2,5-二羟基-1,4-苯二羧醛和2,4,6-三(4-氨基苯基)-1,3,5-三嗪为构筑单元。
优选的,所述fe3o4@sio2-nh2纳米微球的粒径为660~700nm。
优选的,所述壳的厚度为60~120nm。
本发明提供了上述技术方案所述磁性共价有机框架材料的制备方法,包括以下步骤:
(1)将fe3o4@sio2-nh2的1,4-二氧六环分散液和2,5-二羟基-1,4-苯二羧醛的1,4-二氧六环溶液混合进行第一希夫碱反应,得到醛基化磁珠;
(2)将所述醛基化磁珠分散于1,4-二氧六环中后与2,4,6-三(4-氨基苯基)-1,3,5-三嗪的1,4-二氧六环溶液混合,再加入2,5-二羟基-1,4-苯二羧醛的1,4-二氧六环溶液进行第二席夫碱反应,得到磁性共价有机框架材料。
优选的,所述步骤(1)中,以fe3o4@sio2-nh2和2,5-二羟基-1,4-苯二羧醛的量计,所述fe3o4@sio2-nh2溶液的质量和2,5-二羟基-1,4-苯二羧醛的1,4-二氧六环溶液的物质的量之比为(2~3)g:(0.06~0.09)mmol。
优选的,所述步骤(1)中,第一希夫碱反应的温度为50~60℃,时间为1~2h。
优选的,所述步骤(2)中,以fe3o4@sio2-nh2、2,4,6-三(4-氨基苯基)-1,3,5-三嗪和2,5-二羟基-1,4-苯二羧醛的量计,所述醛基化磁珠的质量、2,4,6-三(4-氨基苯基)-1,3,5-三嗪的1,4-二氧六环溶液的物质的量和2,5-二羟基-1,4-苯二羧醛的1,4-二氧六环溶液的物质的量的比为(2~3)g:(0.04~0.06)mmol:(0.08~0.12)mmol。
优选的,所述步骤(2)中,第二希夫碱反应的时间为2~3天。
本发明还提供了上述技术方案所述磁性共价有机框架材料或上述技术方案所述制备方法得到的磁性共价有机框架材料在去除农药有机污染物中的应用。
本发明提供了一种磁性共价有机框架材料,具有核-壳结构,所述核-壳结构中的核为fe3o4@sio2-nh2纳米微球,壳的化学组成为cof;所述cof以2,5-二羟基-1,4-苯二羧醛(bd)和2,4,6-三(4-氨基苯基)-1,3,5-三嗪(ba)为构筑单元。本发明以bd和ba为构筑基元制备的cof保留了现有cof的热稳定性、高比表面积和具有永久孔隙度的优点;同时,bd中的两个羟基与ba中的三嗪环能够提供与农药有机污染物的作用位点且提高了cof的亲水性,使磁性共价有机框架材料在水体中具有良好的分散性;cof-bdba可以以π-π作用、c-h-π作用与有机磷农药结合,进而达到吸附该类药物的目标;而且,cof中的醛基与fe3o4@sio2-nh2中的氨基形成c=n键,结合力强,磁性共价有机框架材料具有超顺磁性,能够通过外加磁场实现磁性共价有机框架材料与吸附介质的简便、快速、有效分离;本发明提供的磁性共价有机框架材料的饱和磁化强度为66.90emu/g。
本发明提供了上述技术方案所述磁性共价有机框架材料的制备方法,包括以下步骤:(1)将fe3o4@sio2-nh2的1,4-二氧六环分散液和2,5-二羟基-1,4-苯二羧醛的1,4-二氧六环溶液混合进行第一希夫碱反应,得到醛基化磁珠;(2)将醛基化磁珠分散于1,4-二氧六环中后与2,4,6-三(4-氨基苯基)-1,3,5-三嗪的1,4-二氧六环溶液混合,再加入2,5-二羟基-1,4-苯二羧醛的1,4-二氧六环溶液进行第二席夫碱反应,得到磁性共价有机框架材料。本发明提供的制备方法,无需在高压密封的严苛反应条件进行,操作简单,适宜工业化生产。
附图说明
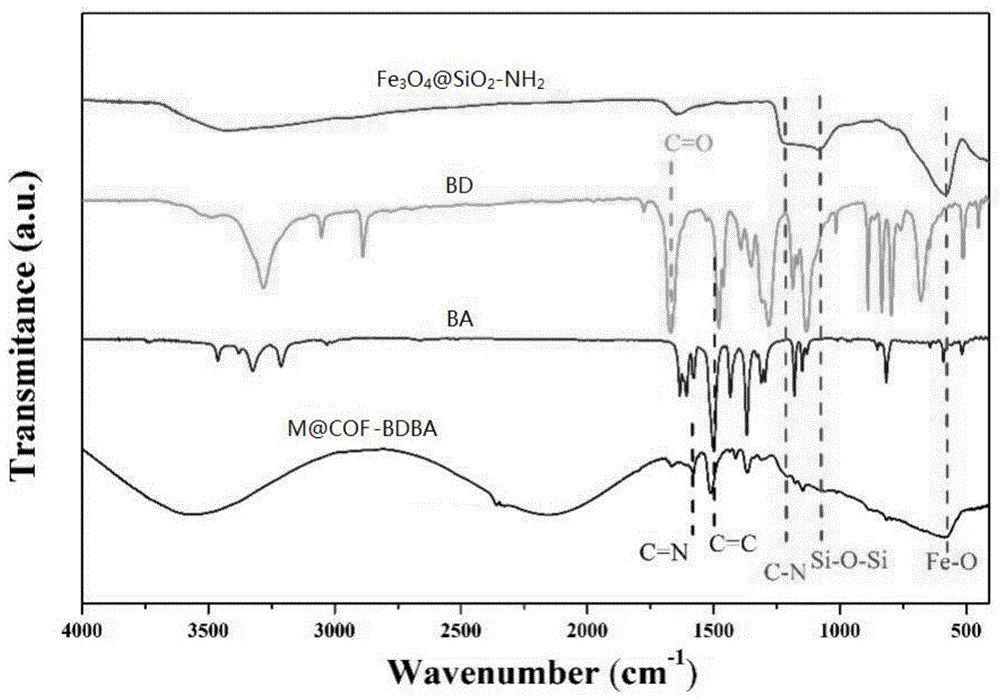
图1为bd、ba、实施例1制备的fe3o4@sio2-nh2和m@cof-bdba的红外谱图;
图2为实施例1制备的fe3o4@sio2-nh2和m@cof-bdba的tem图,其中,a为fe3o4@sio2-nh2,b为m@cof-bdba。
具体实施方式
本发明提供了一种磁性共价有机框架材料,具有核-壳结构,所述核-壳结构中的核的化学组成为fe3o4@sio2-nh2纳米微球,壳的化学组成为cof;所述cof以2,5-二羟基-1,4-苯二羧醛和2,4,6-三(4-氨基苯基)-1,3,5-三嗪为构筑单元。
在本发明中,所述壳的厚度优选为60~120nm,更优选为70~110nm,最优选为80~100nm。
在本发明中,所述fe3o4@sio2-nh2纳米微球的粒径优选为660~700nm,更优选为680~780nm,最优选为700~750nm。
在本发明中,所述fe3o4@sio2-nh2纳米微球的制备方法,优选包括以下步骤:
将磁性fe3o4纳米粒子、碱性试剂、正硅酸乙酯和溶剂混合,进行第一水解缩合反应,得到磁性fe3o4@sio2纳米粒子;
将氨基硅烷偶联剂溶液调节至酸性后加入所述磁性fe3o4@sio2纳米粒子进行第二水解缩合反应,得到fe3o4@sio2-nh2纳米微球。
在本发明中,若无特殊说明,所有的原料组分均为本领域技术人员熟知的市售商品。
在本发明中,所述磁性fe3o4纳米粒子优选自制得到,所述磁性fe3o4纳米粒子的制备方法,优选包括以下步骤:将水溶性铁盐的乙二醇溶液、醋酸钠的乙二醇溶液和柠檬酸的乙二醇溶液混合,依次进行沉淀反应和热分解反应,得到磁性fe3o4纳米粒子。
在本发明中,所述水溶性铁盐的乙二醇溶液的浓度优选为30~60g/l,更优选为40~50g/l;所述水溶性铁盐优选包括氯化铁。在本发明中,所述醋酸钠的乙二醇溶液的浓度优选为50~100g/l,更优选为60~80g/l。在本发明中,所述柠檬酸三钠的乙二醇溶液的浓度优选为4~9g/l,更优选为5.5~8g/l。在本发明中,以水溶性铁盐、醋酸钠和柠檬酸三钠的量计,所述水溶性铁盐的乙二醇溶液、醋酸钠的乙二醇溶液和柠檬酸三钠的乙二醇溶液的质量比优选为1:(3~18):(0.25~1.2),更优选为1:(5~15):(0.5~1)。
在本发明中,所述混合的方式优选为搅拌混合,本发明对于所述搅拌混合的速度和时间没有特殊限定,能够将原料混合均匀即可。在本发明中,所述沉淀反应的温度优选为室温,时间优选为0.5~1h;所述沉淀反应过程中发生的反应为:3ch3coona+fecl3+3h2o=3ch3cooh+fe(oh)3↓+3nacl。所述沉淀反应后,本发明将得到的沉淀体系升温进行热分解反应。在本发明中,所述热分解反应的温度优选为190~220℃,更优选为200~210℃,时间优选为7~12h,更优选为8~10h;所述热分解反应过程中,fe(oh)3发生热分解生成fe3o4,柠檬酸三钠或聚乙二醇与fe3o4形成一层有机膜,通过静电和较小的位阻作用而分散,使fe3o4不易团聚。在本发明中,所述热分解反应的设备优选为带四氟乙烯内衬的反应釜。
所述热分解反应后,本发明优选还包括将所述热分解反应的体系进行磁分离,将所得固体产物依次进行水洗、醇洗和干燥,得到磁性fe3o4纳米粒子。在本发明中,所述水洗的次数优选为3~7次,更优选为4~6次,最优选为5次。在本发明中,所述醇洗优选为无水乙醇洗或无水甲醇洗,所述醇洗的次数优选为3~7次,更优选为4~6次,最优选为5次。在本发明中,所述干燥的温度优选为40~65℃,更优选为45~60℃,最优选为50~55℃;时间优选为8~12h,更优选为9~11h,最优选为10h。在本发明中,所述磁性fe3o4纳米粒子的粒径优选为650~690nm,更优选为660~680nm。
得到磁性fe3o4纳米粒子后,本发明将所述磁性fe3o4纳米粒子、碱性试剂、正硅酸乙酯和溶剂混合,进行第一水解缩合反应,得到磁性fe3o4@sio2纳米粒子。
在本发明中,所述磁性fe3o4纳米粒子和正硅酸乙酯的质量比优选为1:(0.2~1),更优选为1:(0.4~0.8),最优选为1:(0.5~0.6)。在本发明中,所述碱性试剂优选包括氨水,所述氨水的浓度优选为1~3wt%,更优选为1.5~2.5wt%,最优选为2wt%。在本发明中,所述磁性fe3o4纳米粒子和碱性试剂的质量比优选为1:(2~4),更优选为1:(2.5~3.5),最优选为1:3。在本发明中,所述溶剂优选为异丙醇-水混合溶剂,所述混合溶剂中水和异丙醇的体积比优选为1:(4~6),更优选为1:(4.5~5.5),最优选为1:5。在本发明中,所述磁性fe3o4纳米粒子质量和溶剂体积之比优选为1g:(10~15)ml,更优选为1g:(11~14)ml,最优选为1g:(12~13)ml。
在本发明中,所述磁性fe3o4纳米粒子、碱性试剂、正硅酸乙酯和溶剂混合的方式优选为超声混合,本发明对于所述超声混合没有特殊限定,能够将原料混合均匀即可。在本发明中,所述磁性fe3o4纳米粒子、碱性试剂、正硅酸乙酯和溶剂混合的顺序优选为将磁性fe3o4纳米粒子、碱性试剂和溶剂混合,在所得混合液中滴加正硅酸乙酯。本发明对于所述滴加的速度没有特殊限定,匀速逐滴加入即可。
在本发明中,所述第一水解缩合反应的温度优选为室温,时间优选为1~5h,更优选为2~4h,最优选为3h。在本发明中,所述水解缩合反应过程中发生的反应为:si(och2ch3)4+h2o=si(oh)4+c2h5oh,然后si(oh)4与fe3o4发生缩合反应生成fe3o4@sio2,即在四氧化三铁表面包覆二氧化硅层。
所述第一水解缩合反应后,本发明优选还包括将所述第一水解缩合反应的体系进行磁分离,将所得固体产物依次进行水洗、醇洗和干燥,得到磁性fe3o4@sio2纳米粒子。在本发明中,所述水洗的次数优选为3~7次,更优选为4~6次,最优选为5次;所述水洗的目的是除去体系中的碱性试剂和水溶性杂质。在本发明中,所述醇洗优选为无水乙醇洗或无水甲醇洗,所述醇洗的的次数优选为3~7次,更优选为4~6次,最优选为5次。在本发明中,所述干燥的温度优选为35~65℃,更优选为40~60℃,最优选为50~55℃;时间优选为10~15h,更优选为11~14h,最优选为12~13h。在本发明中,所述磁性fe3o4@sio2纳米粒子的粒径优选为660~700nm,更优选为670~690nm。
得到磁性fe3o4@sio2纳米粒子后,本发明将氨基硅烷偶联剂溶液调节至酸性后加入所述磁性fe3o4@sio2纳米粒子进行第二水解缩合反应,得到fe3o4@sio2-nh2纳米微球。
在本发明中,所述氨基硅烷偶联剂溶液的浓度优选为0.01~0.04g/ml,更优选为0.02~0.03g/ml;所述氨基硅烷偶联剂优选包括3-氨基丙基三乙氧基硅烷;所述氨基硅烷偶联剂溶液中的溶剂优选为乙醇溶液,所述乙醇溶液中的乙醇和水的体积比优选为(6~9):1,更优选为(7~8):1。在本发明中,所述磁性fe3o4@sio2纳米粒子和氨基硅烷偶联剂的质量比优选为1:(1~4),更优选为1:(1.5~3.5),最优选为1:(2~3)。在本发明中,所述磁性fe3o4@sio2纳米粒子优选以磁性fe3o4@sio2纳米粒子分散液形式使用,所述磁性fe3o4@sio2纳米粒子分散液中的溶剂优选为无水乙醇,浓度优选为20~35g/l,更优选为25~30g/l。在本发明中,所述氨基硅烷偶联剂的质量和溶剂的体积之比优选为1g:(30~70ml),更优选为1g:(40~50ml)。
在本发明中,所述将氨基硅烷偶联剂和溶剂混合的方式优选为在搅拌条件下将氨基硅烷偶联剂滴加到溶剂中,本发明对于所述搅拌的速度没有特殊限定,能够将原料混合均匀即可;本发明对于所述滴加的速度没有特殊限定,匀速逐滴加入即可。
在本发明中,所述调节ph值至酸性的ph值优选为3.5~4.5,更优选为4;本发明对于所述调节ph值采用的酸没有特殊限定,采用本领域技术人员熟知的弱酸即可,具体如冰醋酸。
在本发明中,所述第二水解缩合反应的温度优选为65~75℃,更优选为70℃,时间优选为3~6h,更优选为4~5h。在本发明中,所述第二水解缩合反应过程中,氨基硅烷偶联剂中的烷氧基水解后可与硅胶表面的羟基反应并将氨基键合到硅胶表面,生成fe3o4@sio2-nh2。
所述第二水解缩合反应后,本发明优选还包括将所述第二水解缩合反应的体系进行磁分离,将所得固体产物依次进行水洗、醇洗和干燥,得到磁性fe3o4@sio2纳米粒子。在本发明中,所述水洗的次数优选为3~7次,更优选为4~6次,最优选为5次;所述水洗的目的是除去体系中的弱酸和水溶性杂质。在本发明中,所述醇洗优选为无水乙醇洗,所述醇洗的的次数优选为3~7次,更优选为4~6次,最优选为5次;所述醇洗的目的是除去未完全反应的原料。在本发明中,所述干燥的温度优选为35~65℃,更优选为40~60℃,最优选为50~55℃;时间优选为10~18h,更优选为12~16h,最优选为14~15h。在本发明中,所述磁性fe3o4@sio2-nh2纳米微球的粒径优选为660~700nm,更优选为670~690nm。
在本发明中,所述2,5-二羟基-1,4-苯二羧醛和2,4,6-三(4-氨基苯基)-1,3,5-三嗪发生席夫碱反应生成cof;cof中的醛基与磁性fe3o4@sio2-nh2纳米微球中的氨基通过含碳氮双键(c=n)的亚胺连接。
本发明提供了上述技术方案所述磁性共价有机框架材料的制备方法,包括以下步骤:
(1)将fe3o4@sio2-nh2的1,4-二氧六环分散液和2,5-二羟基-1,4-苯二羧醛的1,4-二氧六环溶液混合进行第一希夫碱反应,得到醛基化磁珠;
(2)将所述醛基化磁珠分散于1,4-二氧六环中后与2,4,6-三(4-氨基苯基)-1,3,5-三嗪的1,4-二氧六环溶液混合,再加入2,5-二羟基-1,4-苯二羧醛的1,4-二氧六环溶液进行第二席夫碱反应,得到磁性共价有机框架材料。
本发明将fe3o4@sio2-nh2的1,4-二氧六环分散液和2,5-二羟基-1,4-苯二羧醛的1,4-二氧六环溶液混合进行第一希夫碱反应,得到醛基化磁珠。
在本发明中,所述fe3o4@sio2-nh2的1,4-二氧六环分散液的浓度优选为20~30g/l,更优选为22~28g/l,最优选为25g/l。
在本发明中,所述2,5-二羟基-1,4-苯二羧醛的1,4-二氧六环溶液的浓度优选为0.4~0.6mmol/l,更优选为0.45~0.55mmol/l,最优选为0.5mmol/l。
在本发明中,以fe3o4@sio2-nh2和2,5-二羟基-1,4-苯二羧醛的量计,所述fe3o4@sio2-nh2的1,4-二氧六环分散液的质量和2,5-二羟基-1,4-苯二羧醛的1,4-二氧六环溶液的物质的量之比优选为(2~3)g:(0.06~0.09)mmol,更优选为(2.2~2.8)g:(0.065~0.085)mmol,最优选为(2.4~2.6)g:(0.07~0.08)mmol。
在本发明中,所述混合优选为超声混合,本发明对于所述超声混合的功率和时间没有特殊限定,能够混合均匀即可。
在本发明中,所述第一希夫碱反应的温度优选为50~60℃,更优选为52~58℃,最优选为54~56℃;时间优选为1~2h,更优选为1.2h~1.8h,最优选为1.5~1.6h。在本发明中,所述第一希夫碱反应过程中发生的反应如图1所示,fe3o4@sio2-nh2中-nh2和bd中的-c=o缩合生成含碳氮双键(c=n)的亚胺,得到醛基化磁珠。
所述第一希夫碱反应后,本发明优选还包括将所述第一希夫碱反应的体系进行磁分离,将所得固体产物进行有机溶剂洗涤后干燥,得到醛基化磁珠。在本发明中,所述有机溶剂洗涤用的有机溶剂优选为1,4-二氧六环,所述有机溶剂洗涤的次数优选为3~7次,更优选为4~6次,最优选为5次;每次用的有机溶剂的体积优选为20~50ml,更优选为30~40ml。本发明对于所述干燥的温度和时间没有特殊限定,干燥至恒重即可。
得到醛基化磁珠后,本发明将醛基化磁珠分散于1,4-二氧六环中后与2,4,6-三(4-氨基苯基)-1,3,5-三嗪的1,4-二氧六环溶液混合,再加入2,5-二羟基-1,4-苯二羧醛的1,4-二氧六环溶液进行第二席夫碱反应,得到磁性共价有机框架材料。
在本发明中,所述分散的方式优选为超声分散,本发明对于所述超声分散的功率和时间没有特殊限定,能够将醛基化磁珠均匀分散于1,4-二氧六环中即可。在本发明中,所述醛基化磁珠分散于1,4-二氧六环中得到的醛基化磁珠分散液的浓度优选为20~30g/l,更优选为22~28g/l,最优选为25g/l。
在本发明中,所述2,4,6-三(4-氨基苯基)-1,3,5-三嗪的1,4-二氧六环溶液的浓度优选为0.4~0.6mmol/l,更优选为0.45~0.55mmol/l,最优选为0.5mmol/l。
所述2,5-二羟基-1,4-苯二羧醛的1,4-二氧六环溶液的浓度优选为0.4~0.6mmol/l,更优选为0.45~0.55mmol/l,最优选为0.5mmol/l。
在本发明中,以fe3o4@sio2-nh2、2,4,6-三(4-氨基苯基)-1,3,5-三嗪和2,5-二羟基-1,4-苯二羧醛的量计,所述醛基化磁珠的质量、2,4,6-三(4-氨基苯基)-1,3,5-三嗪的1,4-二氧六环溶液的物质的量和2,5-二羟基-1,4-苯二羧醛的1,4-二氧六环溶液的物质的量的比优选为(2~3)g:(0.04~0.06)mmol:(0.08~0.12)mmol,更优选为(2.2~2.8)g:(0.045~0.055)mmol:(0.085~0.11)mmol,最优选为(2.4~2.6)g:(0.045~0.05)mmol:(0.09~0.1)mmol。
在本发明中,所述混合优选为搅拌混合,本发明对于所述搅拌混合的速度和时间没有特殊限定,能够将原料混合均匀即可。在本发明中,所述2,5-二羟基-1,4-苯二羧醛的1,4-二氧六环溶液的加入方式优选为滴加,本发明对于所述滴加的速度没有特殊限定,匀速逐滴加入即可。本发明采用上述加料方式,能够保证反应向生成cof的方向进行。
在本发明中,所述第二希夫碱反应的温度优选为室温;压力优选为常压;时间优选为2~3天,更优选为2.2~2.8天,最优选为2.5天。在本发明中,所述第二希夫碱反应过程中发生的反应如图1所示,醛基化磁珠中的醛基与ba中的氨基发生希夫碱反应,然后与bd中的醛基发生希夫碱反应,在fe3o4@sio2-nh2表面接枝cof,生成磁性共价有机框架材料(简称m@cof-bdba,其中,m表示fe3o4@sio2-nh2)。
所述第二希夫碱反应后,本发明优选还包括将所述第二希夫碱反应的体系进行磁分离,将所得固体产物进行有机溶剂洗涤后干燥,得到磁性共价有机框架材料。在本发明中,所述洗涤用的溶剂优选为乙醇或水,所述洗涤优选为超声洗涤,所述超声洗涤的时间优选为3~5min,更优选为4min;本发明对于所述超声洗涤的功率没有特殊限定,能够将磁性共价有机框架材料表面的杂质去除干净即可。在本发明中,所述洗涤的次数优选为3~7次,更优选为4~6次,最优选为5次;每次洗涤用的溶剂的体积优选为50~130ml,更优选为60~120ml,最优选为80~100ml。在本发明中,所述干燥的温度优选为35~65℃,更优选为40~60℃,最优选为50~55℃;时间优选为10~18h,更优选为12~16h,最优选为14~15h。
本发明在常温常压下即可制备的磁性共价有机框架材料,改进了cof材料传统合成方法中高温高压密闭反应的严苛条件,避免了液氮闪冻、火焰密封等步骤,反应条件温和,操作简单;而且,已报道的cof材料大都是低密度的粉末状固体,质量较轻,即便通过离心等操作的辅助,也难以保证材料的高效快速分离和回收,而本发明提供的制备方法,能够使得cof在fe3o4表面生长,从一定程度上支撑了cof的形态,改善了cof堆积在一起的形态,本发明制备的m@cof-bdba能够在去除农药有机污染物后通过简单的外加磁铁而被分离回收,整个过程无需利用离心机辅助,简化了操作,节省了时间,快速、便捷、高效,适宜工业化生产。
本发明还提供了上述技术方案所述磁性共价有机框架材料或上述技术方案所述制备方法得到的磁性共价有机框架材料在去除农药有机污染物中的应用。
在本发明中,所述应用优选为去除水体中农药有机污染物的应用。本发明对于所述农药有机污染物的种类没有特殊限定,所述农药有机污染物包括含有苯环、氨基、羟基中的一种或几种基团即可,具体如有机磷,所述有机磷优选包括辛硫磷、哒嗪硫磷、o,o-二乙基-s-(乙硫基甲基)二硫化磷酸酯(甲拌磷)和嘧啶磷中的一种或几种。
在本发明中,所述磁性共价有机框架材料应用后的回收方法,优选包括以下步骤:通过外加磁铁将应用体系中的磁性共价有机框架材料分离,并进行洗涤后干燥,得到回收磁性共价有机框架材料。在本发明中,所述洗涤用的溶剂优选为乙醇或水,所述洗涤优选为超声洗涤,所述超声洗涤的时间优选为3~5min,更优选为4min;本发明对于所述超声洗涤的功率没有特殊限定,能够将磁性共价有机框架材料表面的杂质去除干净即可。在本发明中,所述洗涤的次数优选为3~7次,更优选为4~6次,最优选为5次;每次洗涤用的溶剂的体积优选为50~130ml,更优选为60~120ml,最优选为80~100ml。在本发明中,所述干燥的温度优选为35~65℃,更优选为40~60℃,最优选为50~55℃;时间优选为10~18h,更优选为12~16h,最优选为14~15h。
下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例1
在搅拌条件下,将16ml浓度为40g/l的fecl3·6h2o乙二醇溶液、60ml浓度为80g/l的ch3coona·3h2o乙二醇溶液与60ml浓度为5.5g/l的na3c6h5o7·2h2o乙二醇溶液混合均匀,在室温下沉淀反应0.5h,然后将所得反应体系移至聚四氟乙烯内衬反应釜中,在210℃热分解反应10h,外加磁铁吸附分离,将所得固体产物依次进行水洗5次,无水乙醇洗5次,在65℃干燥10h,得到粒径为650~690nm的磁性fe3o4纳米粒子;
将2g磁性fe3o4纳米粒子、25ml水、120ml异丙醇和3ml浓度为2wt%的氨水超声混合,匀速逐滴加入0.5ml正硅酸乙酯,在搅拌、室温下进行第一水解缩合反应2h,外加磁铁吸附分离,将所得固体产物依次进行水洗5次,无水乙醇洗5次,在65℃干燥10h,得到粒径为660~700nm的磁性fe3o4@sio2纳米粒子;
在90ml乙醇水溶液(乙醇:水体积比=8:1)中匀速逐滴加入2ml3-氨基丙基三乙氧基硅烷,用冰醋酸调至ph值至4,加入30ml浓度为30g/l的fe3o4@sio2的无水乙醇溶液,在70℃、搅拌条件下进行第二水解缩合反应4h。外加磁铁分离,将所得固体产物依次进行水洗5次,无水乙醇洗5次,在65℃干燥12h,得到粒径为660~700nm的fe3o4@sio2-nh2。
将30ml浓度为0.5mmol/l的bd的1,4-二氧六环溶液与20ml浓度为25g/l的fe3o4@sio2-nh2的1,4-二氧六环溶液超声混匀在50℃、搅拌条件下进行第一希夫碱反应1h,外加磁铁磁分离,将所得固体产物用1,4-二氧六环洗涤3次,每次洗涤1,4-二氧六环的用量为30ml,在65℃干燥12h,得到醛基化磁珠;
将所述醛基化磁珠加入至20ml1,4-二氧六环中超声分散,加入20ml浓度为0.5mmol/l的ba的1,4-二氧六环溶液搅拌混合均匀,逐滴加入40ml0.5mmol/l的bd的1,4-二氧六环溶液搅拌混合均匀,在常温、常压下第二席夫碱反应3天,磁分离,将所得固体产物用乙醇洗涤4次,每次洗涤乙醇的用量为100ml,在65℃干燥12h,得到粒径为760~800nm的磁性共价有机框架材料(简称m@cof-bdba)。
bd、ba、本实施例制备的fe3o4@sio2-nh2和m@cof-bdba的红外谱图如图1所示。由图1可知,fe3o4@sio2-nh2出现fe-o、si-o-si、和c-n特征峰,说明成功制备得到fe3o4@sio2-nh2;m@cof-bdba中除出现fe-o、si-o-si、和c-n三个特征峰外,还具有bd和ba的c=n和c=c特征峰,说明bd和ba成功接枝到fe3o4@sio2-nh2表面,但由于fe3o4@sio2-nh2被cof层包裹,fe-o、si-o-si、和c-n三个特征峰的强度相对减弱。
本实施例制备的fe3o4@sio2-nh2和m@cof-bdba的tem图如图2所示,其中,a为fe3o4@sio2-nh2,b为m@cof-bdba。由图2可知,fe3o4@sio2-nh2的直径为660~700nm,而m@cof-bdba的外围新增100nm左右的cof膜,m@cof-bdba的直径为760~800nm。
应用例1
吸附动力学研究
在40ml混合有机磷水样中加入40mg实施例1制备的m@cof-bdba,置于25℃恒温摇床上震荡,分别在0min、10min、20min、40min、60min和100min时取样,用0.22μm的微孔过滤膜过滤后,用lc-ms/ms测定农药水样中的有机磷浓度,其中,有机磷为哒嗪硫磷(pyridafenthion)、甲拌磷(phorate)、辛硫磷(phoxim)或嘧啶磷(pyrimitate),4种有机磷的初始浓度均为2mg/l。
吸附容量计算公式如式(1)所示:
式(1),qe为吸附平衡时m@cof-bdba的吸附容量,单位为mg/g;c0为有机磷的初始浓度,单位为mg/l;ce为吸附平衡时有机磷的浓度,单位为mg/l,v为农药水样的体积,单位为l;m为m@cof-bdba的质量,单位为g。
表1不同吸附时间下水样中有机磷的浓度和吸附容量
由表1可知,m@cof-bdba对哒嗪硫磷(pyridafenthion)、甲拌磷(phorate)、辛硫磷(phoxim)、嘧啶磷(pyrimitate)的吸附速率都很快,致使水样中的有机磷浓度迅速下降,10min即可达到吸附平衡,吸附平衡时间短,吸附效率高。
应用例2
(1)在蒸馏水中加入辛硫磷、哒嗪硫磷、甲拌磷和嘧啶磷,得到混合有机磷纯水溶液,其中,4种有机磷的浓度相同,均为5μg/l、10μg/l或50μg/l。
(2)将购买的海盐配制成质量浓度为3%的海盐水溶液作为模拟海水,加入辛硫磷、哒嗪硫磷、甲拌磷和嘧啶磷,得到混合有机磷海水溶液,其中,4种有机磷的浓度相同,均为5μg/l、10μg/l或10μg/l。
(3)分别取40ml所述混合有机磷海水溶液和混合有机磷纯水溶液置于锥形瓶中,每档浓度平行3次实验,共计18个有机磷样品溶液,分别加入40mg实施例1制备的m@cof-bdba,置于25℃恒温摇床上震荡10min后,通过外加磁铁使得m@cof-bdba被吸附在锥形瓶壁,取上层清液过0.22μm微孔过滤膜后,用lc-ms/ms测定样品溶液中的有机磷的浓度,根据式(2)计算有机磷的移除率,结果表2所示。
式(2)中,c0为有机磷的初始浓度,单位为mg/l;ce为吸附平衡时有机磷的浓度,单位为mg/l,removal为有机磷的移除率,单位为%。
表2实施例1制备的m@cof-bdba混合有机磷海水溶液和混合有机磷纯水溶液中有机磷的吸附效果
由表2可知,实施例1制备的m@cof-bdba对蒸馏水中4种有机磷的吸附率大于90.68%,对模拟海水溶液中4种有机磷吸附率大于81.27%,表明,本发明制备的m@cof-bdba对辛硫磷、哒嗪硫磷、甲拌磷和嘧啶磷4种有机磷农药的去除效果优异。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!