一种天然产物ScleropentasideA的合成方法与流程
一种天然产物scleropentaside a的合成方法
技术领域
[0001]
本发明涉及一种天然药物的合成方法,特别涉及一种天然scleropentaside a 的人工合成方法,属于药物合成技术领域。
背景技术:
[0002]
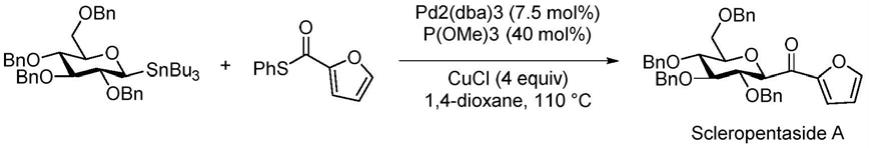
scleropentasides是一类非常经典的天然产物,它广泛存在于植物 scleropyrum pentadrum和植物dendrotrophe frutescens的根茎和叶子中,这些植物在亚洲传统医学中可以被用来避孕、治疗风湿性病痛和皮肤疾病等。因此,scleropentasides是一类具有非常优异医疗价值的天然产物。并且,scleropentasides 在化学结构上也具有于其他天然糖类化合物不同的表现,在许多天然产物糖类化合物中,异头位的c-苷构型一般表现为α构型,而scleropentasides类化合物的糖环上的异头位表现为β构型。在2012年,tripetch kanchanapoom,wannaporndisadee等人在杂志《phytochemistry》上发表的文章unprecedentedfuran-2-carbonyl c-glycosides and phenolic diglycosides from scleropyrumpentandrum中对scleropentasides类化合物做了详细的结构表征。在2018年, maciej a.walczak,feng zhu,jacob rodriguez和sloane o’neill在发表的文章 acyl glycosides through stereospecific glycosyl cross-coupling:rapid access toc(sp3)linked glycomimetics中提到了天然产物scleropentaside a的合成,如图1 所示,全苄基保护的scleropentaside a被合成,产率只有40%。
[0003][0004]
在2019年,g.jacob boehlich和nina schgtzenmeister发表文章β-selective c-glycosylation and its application in the synthesis of scleropentaside a中报道了天然产物scleropentaside a的全合成方案:
[0005][0006]
如图2所示,从最开始的底物到最终的天然产物scleropentaside a的合成共需要 4~5步,并且,总产率只有47%。
[0007]
因此,发明一种高效、简单的天然产物scleropentaside a合成方法线的尤为必要。
技术实现要素:
[0008][0009]
针对现有技术中对于天然产物scleropentaside a的合成方法存在收率较低等缺
陷,本发明的目的是在于提供一种以糖基酸为底物的天然产物scleropentasidea的合成方法,与现有的合成方法相比,在产率方面具有很大的提升,总产率达到69%以上,并且可以做到克级别的产量,且采用的试剂对人体安全。
[0010]
为了实现上述技术目的,本发明提供了一种天然产物scleropentaside a的合成方法,该方法包括以下步骤:
[0011]
1)糖基酸化合物与硫代苯酚进行酯化反应,得到硫代糖酸酯化合物;
[0012]
2)所述硫代糖酸酯化合物与2-呋喃硼酸进行偶联-脱硫反应,得到糖基呋喃酮化合物;
[0013]
3)所述糖基呋喃酮化合物与甲醇进行羰基保护反应,得到缩酮化合物;
[0014]
4)所述缩酮化合物进行脱保护基反应,即得;
[0015]
所述糖基酸化合物具有式1结构:
[0016][0017]
所述硫代糖酸酯化合物具有式2结构:
[0018][0019][0020]
所述糖基呋喃酮化合物具有式3结构:
[0021][0022]
所述糖基呋喃酮化合物具有式4结构:
[0023][0024]
作为一个优选的技术方案,糖基酸化合物与硫代苯酚在hatu缩合试剂和 dipea偶联试剂及有机碱作用下进行酯化反应,得到硫代糖酸酯化合物。酯化反应采用二氯甲烷作为良性反应溶剂。酯化反应过程中先将糖基酸、edci、hatu 和dipea在0℃下搅拌15分钟,充分混合后,再加入硫代苯酚进行反应。
[0025]
作为一个较优选的技术方案,糖基酸化合物与有机碱的摩尔比为1:2~2.3,其中,有机碱为dipea。
[0026]
作为一个较优选的技术方案,糖基酸化合物与hatu缩合试剂和dipea偶联试剂的摩尔比为1:4~4.2:4~4.2。
[0027]
作为一个较优选的技术方案,所述酯化反应的条件为:在室温下反应6~10 小时。
[0028]
在本发明优选的反应条件下,糖基酸化合物与硫代苯酚进行酯化反应得到硫代糖酸酯化合物的收率达到96%以上。
[0029]
作为一个优选的技术方案,所述硫代糖酸酯化合物与2-呋喃硼酸在cutc和 pd2(dba)3催化剂及p(oet)3脱硫剂作用下进行偶联-脱硫反应,得到糖基呋喃酮化合物。偶联-脱硫反应采用四氢呋喃作为反应溶剂。
[0030]
作为一个较优选的技术方案,硫代糖酸酯化合物与2-呋喃硼酸的摩尔比为 0.16:0.27~0.3。
[0031]
作为一个较优选的技术方案,硫代糖酸酯化合物与cutc催化剂及pd2(dba)3催化剂的摩尔比为0.16:0.27~0.3:0.003~0.0035。
[0032]
作为一个较优选的技术方案,硫代糖酸酯化合物与p(oet)3脱硫剂的摩尔比为0.16:0.03~0.035。
[0033]
作为一个较优选的技术方案,所述偶联-脱硫反应的条件为:在氮气保护下,在室温下,反应6~16小时。
[0034]
在本发明优选的反应条件下,硫代糖酸酯化合物与2-呋喃硼酸进行偶联-脱硫反应,得到糖基呋喃酮化合物的收率达到96%以上。
[0035]
作为一个优选的技术方案,所述糖基呋喃酮化合物与甲醇在脱水剂及无机酸催化作用下进行缩合反应,得到缩酮化合物。无机酸优选为硫酸,其加入量为催化量。缩合反应过程中甲醇同时作为羰基保护试剂和溶剂,其加入量相对糖基呋喃酮化合物大大过量。本发明采用的甲醇作为保护基团,其常见易得,分子量小,易操作,且与脱水剂原甲酸三甲酯匹配。
[0036]
作为一个较优选的技术方案,糖基呋喃酮化合物与脱水剂的摩尔比为 1:20~21;所述脱水剂为三甲氧基甲烷。
[0037]
作为一个较优选的技术方案,所述缩合反应的条件为:在室温下,反应6~10 小时。
[0038]
本发明通过缩合反应得到的缩酮化合物相对不稳定,容易分解回到糖基呋喃酮,所以需要快速用于下一步脱保护基反应。
[0039]
作为一个优选的技术方案,所述缩酮化合物与甲醇钠在酸性条件下进行脱保护基反应,即得scleropentaside a。脱保护基反应过程中将保护基脱除,同时将特戊酰基脱除(piv)。
[0040]
作为一个较优选的技术方案,缩酮化合物与meona的摩尔比为 0.84:0.25~0.27;
[0041]
作为一个优选的技术方案,所述酸性条件为ph=1.8~2.2,所述酸性条件采用酸性树脂调节。酸性树脂具体如钠型阳离子交换树脂。
[0042]
作为一个优选的技术方案,所述脱保护基反应的条件为:在室温下反应3~5 个小时。
[0043]
在本发明优选的条件下,先将羰基采用甲氧基保护,再水解脱除特戊酰基,可以将糖基呋喃酮化合物转化成scleropentaside a的收率提高至75%以上。
[0044]
本发明的一种天然产物scleropentaside a的合成路线如下:(其中,化合物3 为2-呋喃硼酸)
[0045][0046]
相对现有技术,本发明技术方案带来的有益技术效果:
[0047]
与之前的合成方法相比,本发明方案中所采用的方法对天然产物 scleropentaside a的收率方面具有很大的提升,整个合成过程的总产率达到69%以上,并且可以做到克级别的产量。
[0048]
在现有技术中,在合成天然产物scleropentaside a过程中还会用到有毒、难以获得的化学试剂,而本发明技术方案所用的试剂均无毒,易获得。
[0049]
在现有技术中在脱除保护基的过程中一般的方法采用i2和氧化剂h2o2,在药物合成中,碘试剂的残留对人体具有一定程度危害,过氧化氢在处理方面也有一定条件要求,而本发明技术方案所采用的脱出保护基的方法简单易操作,且试剂均常见且无危害。
附图说明
[0050]
图1为化合物1的核磁氢谱图;
[0051]
图2为化合物1的核磁碳谱图;
[0052]
图3为化合物2的核磁氢谱图;
[0053]
图4为化合物2的核磁碳谱图;
[0054]
图5为化合物4的核磁氢谱图;
[0055]
图6为化合物4的核磁碳谱图;
[0056]
图7为化合物6的核磁氢谱图;
[0057]
图8为化合物6的核磁碳谱图;
[0058]
图9为化合物7的核磁氢谱图;
[0059]
图10为化合物7的核磁碳谱图;
[0060]
图11为化合物4的单晶衍射。
具体实施方式
[0061]
以下实施例旨在进一步说明本发明内容,而不是限制权利要求的保护范围。
[0062]
实施例1
[0063]
步骤一:在干燥的圆底烧瓶中加入糖基酸1(500mg,0.92mmol),edci(352.3 mg,1.84mmol),hatu(1.40g,3.7mmol)and dipea(478.3mg,3.7mmol),然后加入干燥的二氯甲烷(4ml)做为溶剂,在0℃下搅拌15分钟之后加入thiophenol (407.6mg,3.7mmol),待原料糖基酸1消耗完全之后,加入水和乙酸乙酯萃取三次。得到有机相用nacl水溶液洗,然后再
将有机相用无水硫酸钠干燥,旋干溶剂,得到粗产物。将粗产物用柱层析分离提纯,洗脱剂为ea/pe:1:20,可得干净的白色固体产物硫代糖酸酯2(96%,562.0mg).
[0064]
(2s,3r,4s,5r,6r)-2-((phenylthio)carbonyl)-6-((pivaloyloxy)methyl)tetrahyd ro-2h-pyran-3,4,5-triyl tris(2,2-dimethylpropanoate)2;
[0065]
j=12.5,2.0hz,1h),4.24
–
4.04(m,2h),3.82(ddd,j= 10.0,4.5,2.0hz,1h),1.27(s,9h),1.16(s,9h),1.13(s,9h),1.12(s,9h);
13
c nmr (101mhz,cdcl3)δ194.3,178.2,177.2,176.8,176.4,135.0,129.7,129.4,126.5, 81.9,76.4,72.9,69.5,67.5,61.5,39.1,38.9,38.9,38.9,27.3,27.2;hrms(esi)m/z calcd for c
32
h
47
no
10
sna
+
(m+na)
+
660.28129,found 659.28625.
[0066]
步骤二:在n2保护的条件下往干燥的反应瓶中加入硫代糖酸酯2(102.0mg, 0.16mmol),2-呋喃硼酸3(30.4mg,0.27mmol),cutc(51.5mg,0.27mmol), pd2(dba)3(3.0mg,0.003mmol)and p(oet)3(5.3mg,0.03mmol),再加入干燥的四氢呋喃(2ml)作为溶剂,反应体系在25℃下过夜反应。之后往体系中加入2ml 氨水溶液淬灭反应之后加入乙酸乙酯(200ml)萃取,将得到的有机相用饱和 nacl水溶液清洗之后用无水硫酸钠干燥,旋干溶剂,再用柱层析分离提纯,洗脱剂为ea/pe:1:10。可得到糖基呋喃酮4(96%,92.0mg)。
[0067]
(2s,3r,4s,5r,6r)-2-(furan-2-carbonyl)-6-((pivaloyloxy)methyl) tetrahydro-2h-pyran-3,4,5-triyltris(2,2-dimethylpropanoate)23;
[0068]
5.23(t,j=9.6hz,1h),4.50
–
4.37(m,1h),4.17(td,j=12.4,3.3hz,2h),3.84(ddd, j=10.2,4.6,2.0hz,1h),1.20(s,9h),1.14(s,9h),1.09(s,9h),1.03(s,9h);
13
c nmr(101mhz,cdcl3)δ181.3,177.9,177.2,176.4,176.3,150.2,147.7,120.9, 112.4,80.0,76.7,73.2,69.2,67.4,61.8,38.8,38.8,38.7,38.6,27.2,27.1,27.5,26.9; hrms(esi)m/z calcd for c
31
h
46
o
11
na
+
(m+na)
+
617.29323,found 617.29370.
[0069]
在完成步骤二之后,下一步是脱掉糖环上的保护剂(piv)得到天然产物 scleropentaside a,但在这里出现了意料之外的情况,大量实验表明,采用现有技术中多种脱除保护基团的方法,都难以获得高纯的scleropentaside a,有副产物残留,主要是由于化合物4在碱性条件下非常不稳定,常用的碱性条件下脱除保护基的方法在这里并不适用,而是会生成消除的副产物7。
[0070]
以下对照实验组:
[0071][0072][0073]
通过上述实验组可以看出,现有常见脱保护基团的方法,都会产生副产物7。
[0074]
本发明技术方案设计了羰基保护方案,通过采用甲醇将羰基转化成缩酮,减少了邻位羟基的消除活性,从而有效防止消除反应的发生,得到的几乎是100%的scleropentaside a,具体反应条件如下图:
[0075][0076]
步骤三:在n2保护的条件下往干燥的反应瓶中加入糖基呋喃酮4(50.0mg, 0.084mmol),三甲氧基甲烷(1.68mmol)和h2so4(0.84ul)。之后,再往反应体系中加入2ml干燥的meoh,反应在25℃下直至原料4消耗完毕。之后,往体系中加入5ml水和20ml ea萃取,将得到的有机相用饱和nacl水溶液清洗之后再用无水硫酸钠干燥,旋干溶剂,用柱层析分离提纯,洗脱剂为:ea/pe=1:15,产物为不稳定的黄色的油状液体缩酮5。由于缩酮5非常不稳定,容易分解回到糖基呋喃酮4,所以需要直接快速用于下一步反应。
[0077]
步骤四:将上一步得到的产物溶解在2ml甲醇中,再加入meona(13.6mg, 0.25mmol),在室温下反应4个小时,待原料反应完全之后,加入酸性树脂将体系ph值调至2。然后将树脂过滤,用50ml甲醇清洗三次之后,再用柱层析分离提纯,洗脱剂为meoh/dcm:1:5。产物为白色固体(两步产率75%,16.3mg). scleropentaside a 6。
[0078]
9.5hz,1h),3.65(dd,j=12.2,4.6hz,1h),3.32
–
3.24(m,4h),3.12(td,j=9.1,4.4 hz,1h);
13
c nmr(101mhz,dmso-d6)δ184.5,151.4,148.6,121.3,112.7,81.6, 79.6,78.0,71.6,70.0,61.1;hrms(esi)m/z calcd for c
11
h
14
o7na
+
(m+na)
+ 281.06317,found 281.06323.
[0079]
对照实验组:将糖基呋喃酮4直接进行脱保护基,得到的几乎也全部是消除产物,具体步骤如下:
[0080]
在n2保护下,往干燥的反应瓶中加入糖基呋喃酮4(237.6mg,0.4mmol)和 naome(88.6mg,1.64mmol),然后加入2mlmeoh作为溶剂,在室温下搅拌4 个小时。待原料反应完全之后,加入酸性树脂将体系ph值调至2。然后将树脂过滤,用50ml甲醇清洗三次之后,再用柱层析分离提纯,洗脱剂为meoh/dcm: 1:5,产物为消除的副产物7(两步产率98%,94.0mg)。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1