一种磷酸特地唑胺杂质及其制备方法和用途与流程
本发明涉及一种磷酸特地唑胺杂质及其制备方法和用途,属于化学制药领域。
背景技术:
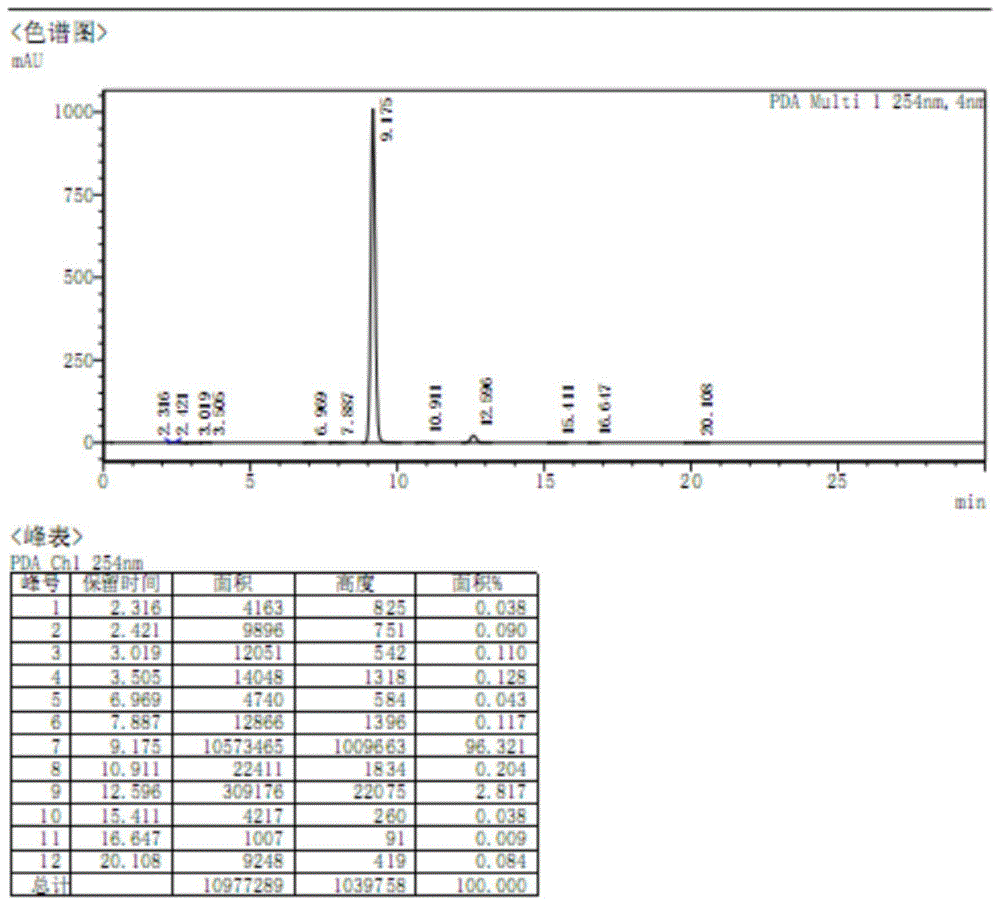
:磷酸特地唑胺是由cubist制药公司研发的一种新型噁唑烷酮类抗菌素,化学名为{(5r)-(3-{3-氟-4-[6-(2-甲基-2h-四唑-5-基)-吡啶-3-基]苯基}-2-氧代噁唑烷-5-基)}甲基磷酸酯。2014年由fda批准上市,临床上用于治疗急性细菌性皮肤和皮肤结构感染。磷酸特地唑胺是一种前药,在体内可被磷酸酶迅速转化为具有生物活性的特地唑胺,从而抑制蛋白质的合成。其作用机制不同于其他抗菌素,不会与其他抗菌素发生交叉耐药现象。临床上主要用于治疗金黄色葡萄球菌(包括耐甲氧西林菌株、甲氧西林敏感菌株)和各种链球菌属和粪肠球菌等革兰氏阳性细菌引起的急性细菌性皮肤和皮肤结构感染。磷酸特地唑胺的化学结构式如下所示:合成磷酸特地唑胺原料药工艺路线中,化合物2-(2-甲基-2h-四氮唑-5-基)-5-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)吡啶是重要的中间体,其化学结构如下:在合成2-(2-甲基-2h-四氮唑-5-基)-5-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)吡啶中间体过程中,会产生杂质,该杂质可参与磷酸特地唑胺原料药合成工艺的后续反应,产生更多的未知杂质,也可单独存在于磷酸特地唑胺原料药中,均严重影响产品质量。但是,现有技术中未见该杂质的公开报道,也没有该杂质的对照品,因此无法在合成磷酸特地唑胺原料药的工艺路线中准确控制该杂质的含量,给磷酸特地唑胺的生产带来安全隐患。杂质在原料药中的存在,可导致制剂的不良反应增加,甚至对患者产生严重后果,因此国际国内药典均对原料药杂质的含量规定了严格的限度。为更好地控制磷酸特地唑胺原料药的产品质量,有必要制备出该杂质对照品,以便在磷酸特地唑胺原料药和相关重要中间体控制其限度。技术实现要素:本发明的目的是提供一种磷酸特地唑胺杂质及其制备方法和用途。本发明提供了一种磷酸特地唑胺杂质,所述杂质的结构式如式i所示:本发明还提供了一种前述的磷酸特地唑胺杂质的制备方法,具体的制备方法包括如下步骤:步骤1:将化合物1、氯化铵和叠氮化钠溶于有机溶剂中,反应,得到化合物2和化合物3的混合物;步骤2:在步骤1得到的化合物2和化合物3的混合物中加入二甲基甲酰胺和碱,抽真空后加入有机溶剂和甲基化试剂,反应,得化合物4和磷酸特地唑胺杂质混合物;步骤3:将化合物4和磷酸特地唑胺杂质混合物溶解于有机溶剂中,加入柱层析硅胶,使用制备液相色谱分离,即得磷酸特地唑胺杂质。进一步地,步骤1中,所述反应为将化合物1、氯化铵和叠氮化钠溶于有机溶剂中,进行第一次反应,再加入氯化铵和叠氮化钠进行第二次反应,即可;优选地,步骤1中,所述第一次反应时,化合物1、氯化铵和叠氮化钠的摩尔比为1:(1~2):(0.8~1.5);所述第二次反应时,化合物1、第二次加入的氯化铵和叠氮化钠的摩尔比为1:(0.15~0.45):(0.1~0.3);所述化合物1与有机溶剂的质量体积比为1g:(8~15)ml;更优选地,步骤1中,所述第一次反应时,化合物1、氯化铵和叠氮化钠的摩尔比为1:(1.5~1.6):(0.98~1.15);所述第二次反应时,化合物1、第二次加入的氯化铵和叠氮化钠的摩尔比为1:(0.28~0.3):(0.223~0.236);所述化合物1与有机溶剂的质量体积比为1g:(10~12.5)ml;进一步优选地,步骤1中,所述第一次反应时,化合物1、氯化铵和叠氮化钠的摩尔比为1:1.5:1.1;所述第二次反应时,化合物1、第二次加入的氯化铵和叠氮化钠的摩尔比为1:0.3:0.2;所述化合物1与有机溶剂的质量体积比为1g:10ml;更进一步优选地,步骤1中,所述有机溶剂为二甲基甲酰胺;和/或,步骤1中,所述第一次反应时,反应温度为70~100℃,反应时间为2~6小时;和/或,步骤1中,所述第二次反应时,反应温度为90~120℃,反应时间为1~3小时;再进一步优选地,步骤1中,所述第一次反应时,反应温度为80~90℃,反应时间为3~4小时;和/或,步骤1中,所述再次反应时,反应温度为100~110℃,反应时间为2~2.5小时。进一步地,步骤2中,化合物2和化合物3的混合物与碱、甲基化试剂质量比为1:(0.25~0.75):(1~2);所述化合物2和化合物3的混合物与二甲基甲酰胺和有机溶剂的质量体积比为1g:(3~6)ml:(4~8)ml;和/或,步骤3中,所述化合物4和磷酸特地唑胺杂质混合物与有机溶剂的质量体积比为1g:(6~15)ml;所述化合物4和磷酸特地唑胺杂质混合物和硅胶的质量比为1:(1~10);优选地,步骤2中,化合物2和化合物3的混合物与碱、甲基化试剂质量比为1:(0.5~0.52):(1.4~1.52);所述化合物2和化合物3的混合物与二甲基甲酰胺和有机溶剂的质量体积比为1g:(4~5)ml:(5~6)ml;和/或,步骤3中,所述化合物4和磷酸特地唑胺杂质混合物与有机溶剂的质量体积比为1g:(10~12)ml;所述化合物4和磷酸特地唑胺杂质混合物和硅胶的质量比为1:3;更优选地,步骤2中,化合物2和化合物3的混合物与碱、甲基化试剂质量比为1:0.5:1.5;所述化合物2和化合物3的混合物与二甲基甲酰胺和有机溶剂的质量体积比为1g:4.5ml:5.5ml;和/或,步骤3中,所述化合物4和磷酸特地唑胺杂质混合物与有机溶剂的质量体积比为1g:10ml;所述化合物4和磷酸特地唑胺杂质混合物和硅胶的质量比为1:3。进一步地,步骤1中,所述反应时加入氮气保护;和/或,步骤2中,所述抽真空为30~50℃搅拌并抽真空3~6小时;和/或,步骤2中,所述碱为氢氧化钠;和/或,步骤2中,所述有机溶剂为四氢呋喃;和/或,步骤2中,所述甲基化试剂为碘甲烷;和/或,步骤2中,所述反应的反应温度为30~60℃,反应时间为1~3小时;和/或,步骤2中,反应后还要提纯,所述提纯步骤如下:反应结束后加入纯水淬灭,然后加入二氯甲烷萃取,浓缩有机相,浓缩物溶于盐酸中,再加入二氯甲烷分液,水相加氢氧化钠至ph>14析出固体,过滤,即得;和/或,步骤3中,所述有机溶剂为二氯甲烷;优选地,和/或,步骤2中,所述抽真空为35~40℃搅拌并抽真空3~6小时;和/或,步骤2中,所述反应的反应温度为40~50℃,反应时间为2~2.5小时。进一步地,所述使用制备液相色谱分离的条件如下:分离参数:淋洗程序:进一步地,使用制备液相色谱分离时,当淋洗液中有杂质2-(2-甲基-四氮唑)-5-叠氮基吡啶峰出现时开始收集,收集至淋洗液中无杂质2-(2-甲基-四氮唑)-5-叠氮基吡啶峰出现时为止,将收集的淋洗液混匀,浓缩至干,即得。本发明还提供了式i所示的磷酸特地唑胺杂质作为对照品在检测磷酸特地唑胺中式i所示的杂质的含量中的用途;本发明还提供了一种磷酸特地唑胺中杂质的检测方法,所述水解杂质为式i所示的杂质,检测方法是高效液相色谱法检测,具体步骤如下:(1)制备供试品溶液,取待测磷酸特地唑胺,溶解,即得;(2)制备对照品溶液,取式i所示的杂质对照品,溶解,即得;(3)分别吸取供试品溶液和对照品溶液注入高效液相色谱仪检测,色谱条件为:色谱柱以十八烷基硅烷键合硅胶为填充剂;流动相由a相和b相组成,a相为磷酸二氢钠水溶液,b相为乙腈;优选地,步骤(1)中,所述供试品溶液中,待测测磷酸特地唑胺浓度为0.25~5g/ml;和/或,步骤(1)中,所述溶解待测磷酸特地唑胺的试剂为磷酸二氢钠水溶液和乙腈等体积混合溶液;和/或,步骤(2)中,所述对照品溶液中,式i所示的杂质对照品的浓度为0.5~5g/ml;和/或,步骤(2)中,所述溶解杂质对照品的试剂为磷酸二氢钠水溶液和乙腈等体积混合溶液;和/或,步骤(3)中,所述高效液相色谱仪检测时,检测波长为280~300nm;和/或,流速为0.8~2ml/min;和/或,柱温为30~40℃;和/或,进样量为10~30μl;更优选地,所述磷酸二氢钠水溶液浓度为0.05~0.15mol/l。进一步地,所述高效液相色谱仪检测时,流动相洗脱程序为梯度洗脱程序,梯度洗脱程序如下:时间(min)流动相b(%)流动相a(%)0.01090102080303070405050414060501585。本发明有关2-(2-甲基-四氮唑)-5-叠氮基吡啶及其制备方法,与现有技术相比,具有以下优点:(1)本发明首次发现磷酸特地唑胺原料药杂质2-(2-甲基-四氮唑)-5-叠氮基吡啶,及首次公开其合成及分离方法;(2)本发明合成步骤为两步,工艺简单、稳定,重复性好;(3)本发明所得杂质2-(2-甲基-四氮唑)-5-叠氮基吡啶的合成及分离方法的纯度高于96%,符合国家药典和中国药典有关杂质对照品的质量要求;(4)本发明制备出合格的2-(2-甲基-四氮唑)-5-叠氮基吡啶的杂质对照品,可应用于磷酸特地唑胺api、原料药和制剂的质量控制,也可为上述工艺研究提供可靠的监测手段,推进工艺优化和确定过程。综上,本发明提供了一种磷酸特地唑胺杂质2-(2-甲基-四氮唑)-5-叠氮基吡啶及其制备方法,该杂质是在合成2-(2-甲基-2h-四氮唑-5-基)-5-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)吡啶中间体过程中产生的杂质,该杂质可参与磷酸特地唑胺原料药合成工艺的后续反应,产生更多的未知杂质,也可单独存在于磷酸特地唑胺原料药中,均严重影响产品质量。本发明将该杂质合成分离出来,且纯度高于96%,符合国家药典和中国药典有关杂质对照品的质量要求。同时,本发明提供了一种检测磷酸特地唑胺中杂质2-(2-甲基-四氮唑)-5-叠氮基吡啶的方法。本发明制备工艺简单、稳定,重复性好,可制备得到杂质2-(2-甲基-四氮唑)-5-叠氮基吡啶对照品,可应用于磷酸特地唑胺api、原料药和制剂的质量控制,也可为上述工艺研究提供可靠的监测手段,推进工艺优化和确定过程,具有良好的应用前景。显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离本发明上述基本技术思想前提下,还可以做出其它多种形式的修改、替换或变更。以下通过实施例形式的具体实施方式,对本发明的上述内容再作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。附图说明图1为实施例1所得产品2-(2-甲基-四氮唑)-5-叠氮基吡啶的hplc图谱。图2为磷酸特地唑胺原料样品中2-(2-甲基-四氮唑)-5-叠氮基吡啶杂质含量测定hplc图谱。具体实施方式本发明具体实施方式中使用的原料、设备均为已知产品,通过购买市售产品获得。部分原料、设备来源如下:(1)原料5-溴-2-氰基吡啶:北京维达化工有限公司生产。柱层析硅胶:西安蓝晓科技新材料股份有限公司生产。磷酸特地唑胺对照品:批号2018a06,宜宾市南溪区红光制药有限公司提供。磷酸特地唑胺原料样品:批号20190506,宜宾市南溪区红光制药有限公司提供。(2)仪器设备高效液相色谱仪:岛津lc-20at;制备液相色谱:博纳艾杰尔agelatechnologies,型号cheetah-mp20。实施例1、磷酸特地唑胺杂质2-(2-甲基-四氮唑)-5-叠氮基吡啶的制备方法(1)2-四氮唑-5-叠氮基吡啶的合成反应式如下:将500ml二甲基甲酰胺(dmf)加入反应瓶,氮气排空(氮气保护),开启搅拌。加入47.5g(0.26mole)5-溴-2-氰基吡啶、21g(0.39mole)氯化铵、19g(0.29mole)叠氮化钠,缓慢升温至80~90℃反应3小时,补加4.2g(0.078mole)氯化铵、3.8g(0.058mole)叠氮化钠100~110℃反应2小时。冷却到室温,过滤得到65.6g2-四氮唑-5-溴吡啶/2-四氮唑-5-叠氮基吡啶混合物,备用,用于下一步投料。(2)2-(2-甲基-四氮唑)-5-叠氮基吡啶的合成反应式如下:将65.6g2-四氮唑-5-溴吡啶/2-四氮唑-5-叠氮基吡啶混合物加入反应瓶中,加入300ml二甲基甲酰胺、33g氢氧化钠,40℃搅拌并抽真空3小时,体系变为白色混浊,测试ph<13且无较大氨气味可进行下一操作。加入360ml四氢呋喃,滴加100g(0.7mole)碘甲烷,滴毕温度保持40~50℃反应2小时,体系溶清,反应结束,加入800ml纯水淬灭反应后400ml二氯甲烷萃取,分出有机相浓缩至干,浓缩物溶于400ml6n盐酸,加入200ml二氯甲烷分液,水相加氢氧化钠至ph>14析出固体,过滤,得2-(2-甲基-四氮唑)-5-叠氮基吡啶/2-(2-甲基-四氮唑)-5-溴吡啶混合物粗品23g。(3)2-(2-甲基-四氮唑)-5-叠氮基吡啶的分离方法取5g的步骤(2)所得2-(2-甲基-四氮唑)-5-叠氮基吡啶/2-(2-甲基-四氮唑)-5-溴吡啶混合物粗品,以50ml二氯甲烷溶解,加15g柱层析硅胶干法制取样品,使用博纳艾杰尔agelatechnologies(型号cheetah-mp2)制备色谱仪分离制备。agelatechnologies(型号cheetah-mp2)参数设置:分离参数:淋洗程序:b%开始结束持续时间当量比8.08.010.018.047.556.4247.550.03.7350.0100.09.9制备液相色谱分离后开始收集,自38号试管开始显示有杂质2-(2-甲基-四氮唑)-5-叠氮基吡啶色谱峰高出现,至80号试管为止,81号试管中已经无杂质2-(2-甲基-四氮唑)-5-叠氮基吡啶色谱峰高,因此收集含有杂质2-(2-甲基-四氮唑)-5-叠氮基吡啶的38~80号试管组分,混合均匀,浓缩至干,得到2-(2-甲基-四氮唑)-5-叠氮基吡啶760mg,测定纯度,纯度为96.3%,符合国家药典和中国药典关于杂质质量标准的要求。2-(2-甲基-四氮唑)-5-叠氮基吡啶的结构式如下:将本实施例所得产品2-(2-甲基-四氮唑)-5-叠氮基吡啶的进行1hnmr图谱测定,解析如下:1hnmr(400mhz,cdcl3):δ8.48(d,j=2.4hz,1h),8.23(d,j=8.5hz,1h),7.51(dd,j=8.5,2.5hz,1h),4.44(s,3h)。将本实施例所的产品2-(2-甲基-四氮唑)-5-叠氮基吡啶进行ms图谱测定,解析如下:ms(es):m/z=203[m+h]+,175[m-n2+h]+。图1所示为实施例1所得产品2-(2-甲基-四氮唑)-5-叠氮基吡啶的hplc纯度测定图谱,纯度为96.3%。实施例2、磷酸特地唑胺杂质2-(2-甲基-四氮唑)-5-叠氮基吡啶的制备方法(1)2-四氮唑-5-叠氮基吡啶的合成反应式如下:将1000ml二甲基甲酰胺(dmf)加入反应瓶,氮气排空(氮气保护),开启搅拌。加入80g5-溴-2-氰基吡啶、38g氯化铵、28g叠氮化钠,缓慢升温至85~90℃反应4小时,补加6.7g氯化铵、6.8g叠氮化钠100~110℃反应2.5小时。冷却到室温,过滤得到112g2-四氮唑-5-溴吡啶/2-四氮唑-5-叠氮基吡啶混合物,备用,用于下一步投料。(2)2-(2-甲基-四氮唑)-5-叠氮基吡啶的合成反应式如下:将100g2-四氮唑-5-溴吡啶/2-四氮唑-5-叠氮基吡啶混合物加入反应瓶中,加入500ml二甲基甲酰胺、52g氢氧化钠,40℃搅拌并抽真空3小时,体系变为白色混浊,测试ph<13且无较大氨气味可进行下一操作。加入600ml四氢呋喃,滴加140g碘甲烷,滴毕温度保持40~45℃反应2.5小时,体系溶清,反应结束,加入1000ml纯水淬灭反应后500ml二氯甲烷萃取,分出有机相浓缩至干,浓缩物溶于500ml6n盐酸,加入250ml二氯甲烷分液,水相加氢氧化钠至ph>14析出固体,过滤,得2-(2-甲基-四氮唑)-5-叠氮基吡啶/2-(2-甲基-四氮唑)-5-溴吡啶混合物粗品36g。(3)2-(2-甲基-四氮唑)-5-叠氮基吡啶的分离方法取5g的步骤(2)所得2-(2-甲基-四氮唑)-5-叠氮基吡啶/2-(2-甲基-四氮唑)-5-溴吡啶混合物粗品,以60ml二氯甲烷溶解,加人15g柱层析硅胶干法制取样品,使用博纳艾杰尔agelatechnologies(型号cheetah-mp2)制备色谱仪分离制备。分离参数和淋洗程序与实施例1相同。有效组分收集的试管编号与实施例1相同,将其浓缩至干,得到2-(2-甲基-四氮唑)-5-叠氮基吡啶772mg,测定纯度,纯度为96.5%,符合国家药典和中国药典关于杂质质量标准的要求。实施例3、将杂质2-(2-甲基-四氮唑)-5-叠氮基吡啶作为对照品,开展磷酸特地唑胺原料质量控制研究(1)稀释液制备:以0.05mol/l磷酸二氢钠水溶液为流动相a,以乙腈为流动相b。取同等体积的a和b混合,摇匀,即为稀释液。(2)精密称取磷酸特地唑胺对照品12.5mg(批号2018a06,)、实施例1制备的2-(2-甲基-四氮唑)-5-叠氮基吡啶对照品25mg,置50ml量瓶中,加入10滴饱和碳酸氢钠和1ml二甲亚砜溶解,再用稀释液稀释至刻度摇匀,作为系统溶液。(3)精密称取磷酸特地唑胺原料药样品12.5mg(批号20190506),置50ml量瓶,加10滴饱和碳酸氢钠溶解,再用稀释液稀释至刻度摇匀,作为供试品溶液。(4)色谱条件以十八烷基硅烷键合硅胶为填充剂,以0.05mol/l磷酸二氢钠水溶液为流动相a,以乙腈为流动相b。流速为0.8ml/min,柱温为30℃,检测波长为300nm,稀释液为a:b(50:50)v/v。梯度洗脱的分配如下表:时间(min)流动相b(%)流动相a(%)0.01090102080303070405050414060501585(5)测定照高效液相色谱法(中国药典2015年版四部通则0512)测定。精密量取稀释液、系统溶液、供试品溶液各10μl注入液相色谱仪,记录色谱图(见图2),供试品溶液色谱图中如有2-(2-甲基-四氮唑)-5-叠氮基吡啶杂质峰,按面积归一化计,空白溶剂峰扣除。(6)测定结果按面积归一化法计算,磷酸特地唑胺原料样品中2-(2-甲基-四氮唑)-5-叠氮基吡啶杂质含量为0.012%。该杂质2-(2-甲基-四氮唑)-5-叠氮基吡啶含量小于0.1%,符合美国药典、欧盟药典、英国药典和中国药典2015年版的质量要求。综上,本发明提供了一种磷酸特地唑胺杂质2-(2-甲基-四氮唑)-5-叠氮基吡啶及其制备方法,该杂质是在合成2-(2-甲基-2h-四氮唑-5-基)-5-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)吡啶中间体过程中产生的杂质,该杂质可参与磷酸特地唑胺原料药合成工艺的后续反应,产生更多的未知杂质,也可单独存在于磷酸特地唑胺原料药中,均严重影响产品质量。本发明将该杂质合成分离出来,且纯度高于96%,符合国家药典和中国药典有关杂质对照品的质量要求。同时,本发明提供了一种检测磷酸特地唑胺中杂质2-(2-甲基-四氮唑)-5-叠氮基吡啶的方法。本发明制备工艺简单、稳定,重复性好,可制备得到杂质2-(2-甲基-四氮唑)-5-叠氮基吡啶对照品,可应用于磷酸特地唑胺api、原料药和制剂的质量控制,也可为上述工艺研究提供可靠的监测手段,推进工艺优化和确定过程,具有良好的应用前景。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1