一种二苯基氧磷取代的菲并咪唑类化合物及其制备方法和应用与流程
本发明涉及有机发光材料及其光电器件应用技术领域,更具体地,涉及一种二苯基氧磷取代的菲并咪唑类化合物及其制备方法和应用。
背景技术:
有机电致荧光材料用于小分子有机电致发光电器件已有很多报道,但大多有机电致荧光材料在聚集态下由于分子的平面结构会形成π-π堆积,导致其荧光被猝灭,降低器件效率。为了提高器件效率,传统的方法会利用基团修饰有机发光材料,如中国专利cn110071220a公开了一种二苯基氧磷修饰的苯并咪唑类材料在oled中的应用,但是其分子自身的电子传输性能一般,导致以其为发光层的器件载流子传输不平衡,器件性能不佳,且由于修饰基团与本体分子间过强的给电子或吸电子能力,导致修饰后的分子具有严重自身分子内电荷转移现象,使其发光红移从而蓝光色纯度不够,影响材料的应用。
因此,寻找一种能同时兼具良好载流子传输平衡能力及高蓝光色纯度的有机电致发光器件材料是本领域技术人员亟待解决的技术问题。
技术实现要素:
本发明要解决的技术问题是克服现有技术的有机电致发光器件材料不能兼具良好载流子传输平衡能力及高蓝光色纯度的缺陷和不足,提供一种二苯基氧磷取代的菲并咪唑类化合物,利用该化合物进一步制得的有机电致发光器件能同时兼具良好载流子传输平衡能力及高蓝光色纯度。
本发明的另一目的是提供上述二苯基氧磷取代的菲并咪唑类化合物的制备方法。
本发明的又一目的是提供上述二苯基氧磷取代的菲并咪唑类化合物的应用。
本发明上述目的通过以下技术方案实现:
一种二苯基氧磷取代的菲并咪唑类化合物,具有式(ⅰ)所示分子结构:
本发明所述化合物具有给体-π-受体结构;其中菲并咪唑单元作为电子给体,二苯基氧磷作为电子受体;可以避免由于过强的电荷转移导致的效率下降现象;本发明所述化合物通过引入具有优秀电荷传输能力的二苯基氧磷基团与菲并咪唑单元配合,有效提高π电子离域,使得电荷的注入和传输更加平衡,具有良好载流子传输平衡能力。并且由于二苯基氧磷的扭曲空间结构,在有机溶剂高效的发射深蓝光,蓝光色纯度高,而且还可以实现固态薄膜聚集态高荧光量子效率;所述化合物可作为发光材料广泛应用于深蓝有机发光器件,也可以作为掺杂器件的主体材料或者是有机发光二极管的电荷传输材料。
本发明还保护上述二苯基氧磷取代的菲并咪唑类化合物的制备方法,包括以下步骤:
s1.将4-溴苯甲醛、双对溴苯胺及9,10-菲醌,通过咪唑成环反应制得4,4-双溴菲并咪唑;
s2.将步骤s1制得的4,4-双溴菲并咪唑和二苯基氧磷通过亲核取代反应制得式(ⅰ)化合物。
优选地,步骤s1所述4-溴苯甲醛、双对溴苯胺及9,10-菲醌的摩尔比为1~2:1~2:2~3。
更优选地,步骤s1所述4-溴苯甲醛、对溴苯胺及9,10-菲醌的摩尔比为1:1:2。
优选地,步骤s1所述咪唑成环反应的温度为115~125℃,反应时间为2~4h。
更优选地,步骤s1反应的温度为120℃,反应时间为2h。
步骤s1的操作可以为,将4-溴苯甲醛、对溴苯胺、9,10-菲醌、乙酸铵混合,再加入冰乙酸,得到黑褐色悬浮液;所述黑色悬浮液在加热搅拌条件下反应至黑褐色变为黑色,并在常温下继续搅拌,分离纯化得4,4-双溴菲并咪唑。其中4,4-双溴菲并咪唑为白色粉末。
优选地,步骤s1的分离操作可以为用甲醇洗涤并过滤,然后真空干燥。
优选地,步骤s1的纯化通过柱层析分离实现柱层析分离时,可以采用硅胶粉作为固定相,可以采用石油醚和二氯甲烷作为洗脱剂。石油醚和二氯甲烷的体积比可以为1∶2。
具体地,步骤s1的反应方程式为:
优选地,步骤s2所述4,4-双溴菲并咪唑和二苯基氧磷的摩尔比为1∶2~3。
更优选地,步骤s2所述4,4-双溴菲并咪唑和二苯基氧磷的摩尔比为1∶2。
优选地,步骤s2中所述亲核取代反应的温度为80~90℃,反应时间为18~36h。
更优选地,步骤s2中所述反应的温度为90℃,反应时间为24h。
优选地,步骤s2的操作可以为,在惰性氛围中,将双溴菲并咪唑、二苯基氧磷硼酸在催化剂氯化镍和锌粉的作用下,与thf和饱和的强碱水溶液混合,加热回流搅拌反应;反应结束后,经萃取、蒸馏、纯化得到式(ⅰ)化合物。式(ⅰ)化合物为黄色粉末。
优选地,步骤s2中萃取可以使用饱和食盐水和二氯甲烷进行;所述蒸馏可以为减压蒸馏。蒸馏后获得黑色固体。
优选地,步骤s2中纯化通过柱层析实现。柱层析纯化时,可以使用硅胶粉作固定相,以石油醚/二氯甲烷为洗脱剂,得到式(ⅰ)化合物。式(ⅰ)化合物为黄色粉末。
步骤s2的反应方程式为:
优选地,步骤s2所述亲核反应的催化剂氯化镍和/或锌粉。
本发明同时保护上述式(ⅰ)化合物作为发光材料在电致发光器件中的应用。
优选地,所述应用包含由所述式(ⅰ)化合物制备的发光层。
与现有技术相比,本发明的有益效果是:
本发明借助菲并咪唑类材料与二苯基氧磷基的优良特性,通过二苯基氧磷基的引入使得载流子注入和传输更加平衡,并且平衡电子给体与受体间的电荷转移作用,实现固态薄膜聚集态高荧光量子效率的同时,还可以保持了其的深蓝发射,合成了兼具良好载流子传输能力、高蓝光色纯度和聚集态下高荧光量子产率的有机发光小分子,其可作为发光材料广泛应用于有机发光器件,特别是稳定高效深蓝有机电致发光器件。同时本发明所述化合物的制备过程简单、便捷、可大规模的批量制备,有利于工厂化的生产制备,有利于其应用的推广。
附图说明
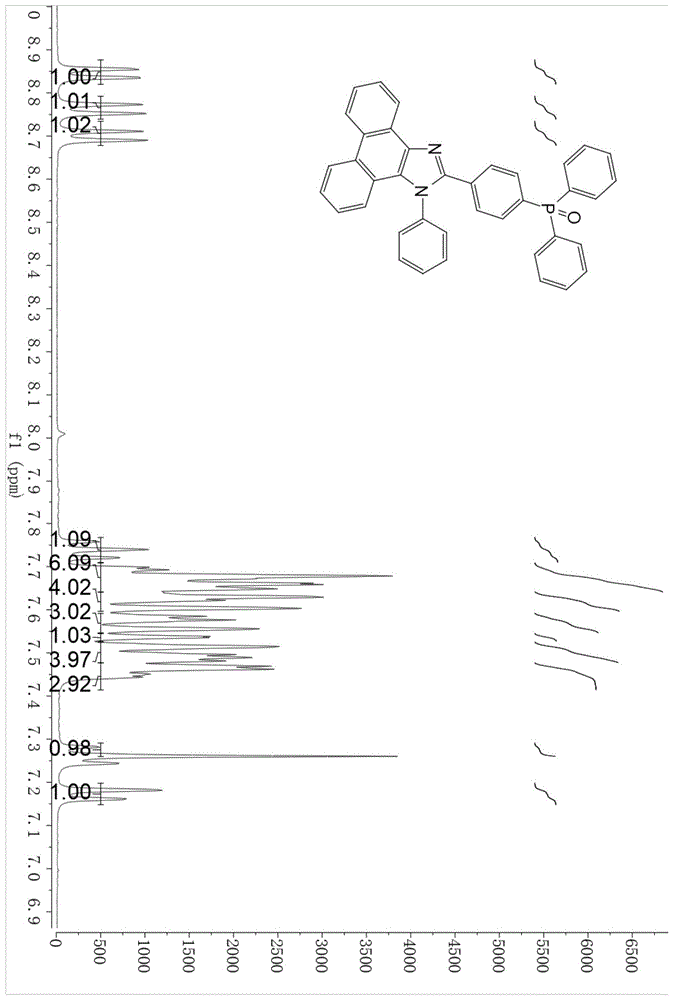
图1为本发明实施例1制得的化合物m1的1hmnr图。
图2为本发明实施例1制得的化合物m1的质谱图。
图3为本发明实施例2制得的化合物m2的1hmnr图。
图4为本发明实施例2制得的化合物m2的质谱图。
图5为本发明实施例1制得的化合物m1和m2在四氢呋喃溶液中的吸收光谱图。
图6为本发明实施例1制得的化合物m1和m2在四氢呋喃溶液中的发射强度图。
图7为本发明实施例1制得的化合物m1和m2在薄膜中的发射光谱图。
具体实施方式
下面结合具体实施方式对本发明作进一步的说明,但实施例并不对本发明做任何形式的限定。除非另有说明,本发明实施例采用的原料试剂为常规购买的原料试剂。
实施例1
一种二苯基氧磷取代的菲并咪唑类化合物,具有式(ⅰ)所示分子结构:
上述二苯基氧磷取代的菲并咪唑类化合物的制备方法,包括如下步骤:
s1.将4-溴苯甲醛(1.86g,10mmol)、对溴苯胺(1.70g,10mmol)、9,10-菲醌(2.08g,10mmol)、乙酸铵(4.62g,60mmol)依次加入100ml的二颈烧瓶(4-溴苯甲醛、双对溴苯胺及9,10-菲醌的摩尔比为1:1:1),并加入60ml的冰乙酸,得到黑褐色悬浮物。混合物在120℃下搅拌2小时后,溶液颜色由黑褐色变为黑色,并在常温下搅拌反应混合物过夜(12小时)。用甲醇洗涤并过滤分离粗产物,然后真空干燥。使用硅胶粉作固定相,石油醚和二氯甲烷为洗脱剂(石油醚:ch2cl2=1:2)纯化产物,得到白色粉末,产率82%。
该反应方程式如下:
s2.将双溴菲并咪唑(1.58g,3mmol)和二苯基氧磷硼酸(1.72g,6mmol),氯化镍(0.16g,0.14mmol),加入100ml的两颈烧瓶中,将烧瓶在真空下抽空并在干燥氮气中置换三次,然后加入60mlthf和加入8ml饱和的k2co3水溶液。在90℃下加热回流搅拌反应48小时。使用饱和食盐水和二氯甲烷萃取。减压蒸馏,获得黑色固体,使用硅胶粉作固定相,以石油醚/二氯甲烷为洗脱剂,通过柱层析法获得1.0g的黄色粉末,为式(ⅰ)化合物,产率40%。
该反应方程式如下:
将步骤s2得到的产物进行减压蒸馏,获得黑色固体,使用硅胶粉作固定相,以石油醚/二氯甲烷为洗脱剂,当洗脱剂体积比例为石油醚:二氯甲烷=2:1时通过柱层析法获得1.0g的白色粉末,为m1化合物,产率40%。
收集完产物m1化合物后,换用洗脱剂体积比例为石油醚:二氯甲烷=1:4时,通过柱层析法获得0.5g的白色粉末,为m2化合物,产率20%。
实施例2
一种二苯基氧磷取代的菲并咪唑类化合物,具有式(ⅰ)所示分子结构:
上述二苯基氧磷取代的菲并咪唑类化合物的制备方法和分离步骤与实施例1相同,区别在于,将步骤s1所述4-溴苯甲醛、双对溴苯胺及9,10-菲醌的摩尔比替换为1:1:2。
结构表征与性能测试
1、测试方法
(1)1hmnr:采用布鲁克400mhz超导核磁共振仪测试氢谱,溶剂为氘代氯仿认。质谱:采用超高效液相色谱串联三重四极杆质谱联用仪,将产物溶于乙腈配成浓度为1mg/ml的溶液进行测试。
(2)在四氢呋喃溶液中的吸收光谱:采用岛津uv-2700紫外可见分光光度计测试,将产物溶解在四氢呋喃溶液,浓度为1×10-5mol/l下的归一化吸收图谱,扫描步长为1nm,停留时间为0.3s,扫描范围为250nm~700nm。
(3)在四氢呋喃溶液中的发射强度:采用爱丁堡fl980瞬态稳态荧光磷光光谱仪在激发波长为320nm下测试所得分子m1及m2在四氢呋喃溶液状态,浓度为1×10-5mol/l下的荧光发射图谱,扫描步长为1nm,停留时间为0.3s,扫描范围为360nm到700nm。
(4)薄膜中的发射光谱:采用爱丁堡fl980瞬态稳态荧光磷光光谱仪在激发波长为320nm下,测试所得分子m1及m2使用真空蒸镀在石英片上的薄膜状态下的荧光发射图谱,扫描步长为1nm,停留时间为0.3s,扫描范围为360nm到700nm。
2、测试结果
m1化合物的核磁图谱如图1所示。从图1可知,其特征波数(ppm)为1hnmr(400mhz,chloroform-d)δ8.85(dd,j=8.0,1.4hz,1h),8.76(d,j=8.3hz,1h),8.70(d,j=8.2hz,1h),7.77–7.71(m,1h),7.71–7.64(m,6h),7.64–7.60(m,4h),7.59–7.55(m,3h),7.54(d,j=1.5hz,1h),7.50(dq,j=10.8,3.2,2.8hz,4h),7.46(dd,j=7.2,3.0hz,3h),7.26(s,1h),7.17(dd,j=8.4,1.3hz,1h).波峰能与菲并咪唑及二苯基氧磷的氢原子一一对应,数量合理,表明合成得到二苯基氧磷取代的菲并咪唑化合物m1,并且结构单一。
m2化合物的核磁图谱如图3所示。从图3可知,其特征波数(ppm)为1hnmr(400mhz,chloroform-d)δ8.85(dd,j=7.9,1.4hz,1h),8.80(d,j=8.4hz,1h),8.73(d,j=8.2hz,1h),7.91(dd,j=11.4,8.2hz,2h),7.79–7.68(m,8h),7.68–7.62(m,9h),7.62–7.57(m,4h),7.56–7.49(m,8h),7.32(td,j=7.6,6.9,1.2hz,1h),7.24–7.19(m,1h).,波峰能与菲并咪唑及二苯基氧磷的氢原子一一对应,数量合理,表明合成得到二苯基氧磷取代的菲并咪唑化合物m2,并且结构单一。
由图2和图4的质谱可知,其碎片峰与所述二苯基氧磷修饰的菲并咪唑化合物m1及m2分子量一致,且无杂峰表明其分子量确定且纯度高。
如图5和图6所示,其中m1及m2分别为单个及双个二苯基氧磷取代的菲并咪唑,箭头指的是曲线读取的纵坐标的方向,采用归一化方法读取吸收强度与发射强度,其最大吸收和发射波长为412nm、426nm,表明其在聚集态下也能高效地发射深蓝荧光,具有高蓝光色纯度,并且聚集状态下其发射峰仍然在420nm左右,显示出其在高效深蓝有机电致发光器件的应用上具有巨大潜力。
图7可以看出,m1及m2分子都具有深蓝光发射,发射波峰都在420nm附近,经测试荧光量子产率可知m1及m2的薄膜荧光量子产率都大于30%,显示其在聚集态的薄膜中也具有高效发光效率,由于膦氧基团自身所具有的高电子迁移率,可见m1及m2分子具有高效载流子运输的性质。
显然,本发明的上述实施例仅仅是为清楚地说明本发明所作的举例,而并非是对本发明的实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明权利要求的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!