一种消旋沙丁胺醇的制备方法与流程
本发明涉及医药化工技术领域,具体地说是一种消旋沙丁胺醇的制备方法。
背景技术:
沙丁胺醇(salbutamol),化学名为1-(4-羟基-3-羟甲基苯基)-2-(叔丁胺基)乙醇,分子式为c13h21no3,分子量为239.31,cas登记号:18559-94-9。沙丁胺醇是由英国葛兰素公司(gsk)开发的强效速效选择性β-受体激动剂,于1968年首次上市,1988年在我国注册,商品名为ventolin。
沙丁胺醇是一种作用被广泛证实的β2受体激动剂,它的作用机制是通过选择性激动β2受体,使支气管平滑肌舒张,对支气管平滑肌中β2受体具有很强的选择性作用,适应症包括:哮喘性支气管炎,支气管哮喘和肺气肿患者的支气管痉挛等呼吸道疾病。该药物以其良好的药效,成为治疗哮喘和慢性阻塞性肺疾病(copd)的首选药物。
沙丁胺醇结构式如下:
现有技术中的消旋沙丁胺醇的合成方法,主要有以下三类:
1、以水杨醛为原料,经傅-克酰基化反应、亚胺合成、亲核取代、水解、钯碳还原得到沙丁胺醇,反应路线如下,
但此路线存在以下缺点:(1)反应原料水杨醛上的酚羟基易氧化和发生亲核取代,使得反应工艺杂质增多;(2)傅-克酰基化反应具有区域选择性,会生成邻位异构体,导致反应收率下降;(3)反应生成亚胺不稳定,使得反应过程不可控,纯度下降。
2、franciscoayala-mata在2012年的molecules中报道了如下的合成路线,
在此种合成方法中,反应路线较长,且其中涉及氢化锂铝还原,正丁基锂拔溴等反应,对反应条件要求苛刻,存在安全隐患,不适合作为工业化生产的反应路线。
3、2020年的tetrahedronletters中报道了如下的合成路线,
此反应过程不需要进行柱层析分离,但使用5-乙酰水杨酸甲酯为原料,价格昂贵,生产成本高,并且在酯的还原时用到红铝,红铝极易吸水,而且淬灭后产生大量的乙二醇单甲醚,增加了三废处理成本,同时产生的偏铝酸钠难以过滤,因此不宜进行大规模工业化生产。
因此,提供一种收率高,原料易得,绿色环保且适合工业化生产要求的消旋沙丁胺醇的制备方法十分有必要。
技术实现要素:
鉴于此,本发明提供了一种消旋沙丁胺醇的制备方法,收率大幅提升,原料价低易得,反应过程中不涉及高危剧毒试剂,后处理时无需柱层析,既适合工业化生产要求又符合“绿色化学”理念。
本发明采用的技术方案是:
一种消旋沙丁胺醇的制备方法,包括以下步骤:
s1、以5-溴水杨醛为原料,与硼氢化钠经还原反应,得到中间体i;
s2、中间体i与氢化钠、卤化苄经烷基化反应,得到中间体ii;
s3、中间体ii与乙烯基三氟硼酸钾经交叉偶联反应,得到中间体iii;
s4、中间体iii与n-溴代琥珀酰亚胺经加成反应,得到中间体iv;
s5、中间体iv与叔丁胺经烷基化反应,得到中间体v;
s6、中间体v在氢气氛围下发生脱保护反应,得到消旋沙丁胺醇。
优选地,
在步骤s1中,将5-溴水杨醛溶于有机溶剂,降温至0~5℃后加入硼氢化钠,然后升至室温,搅拌反应,得到中间体i,反应方程式如下:
中间体i;
本发明首次以价廉易得的5-溴水杨醛为起始反应原料合成消旋沙丁胺醇,使生产成本大大降低;通过溴原子的引入,可使反应在5位上定向引入官能团,提高了反应的选择性和收率;
在步骤s2中,将中间体i溶于有机溶剂,降温至0~5℃加入氢化钠,搅拌均匀后滴加卤化苄,然后升至室温,搅拌反应,得到中间体ii,反应方程式如下:
中间体i中间体ii
其中x为卤素;在这一步中,使用卤化苄对中间体i的羟基进行保护,有效避免了后续工艺过程中副反应的发生,杂质的引入;使用钯碳/h2体系来代替剧毒的肼类化合物进行脱保护,大大降低了反应过程中的污染,符合“绿色化学”理念;
在步骤s3中,将中间体ii溶于溶剂,加入乙烯基三氟硼酸钾、第一钯催化剂和碱,加热回流反应,得到中间体iii,反应方程式如下:
中间体ii中间体iii;
本发明首次将suzuki-miyaura交叉偶联反应用于消旋沙丁胺醇的合成反应中,使用催化量的钯催化剂,便可实现芳环上双键的高效引入,提高反应收率的同时降低了生产成本;
在步骤s4中,将中间体iii溶于溶剂,加入n-溴代琥珀酰亚胺,室温下反应,得到中间体iv,反应方程式如下:
中间体iii中间体iv;
在步骤s5中,将中间体iv和叔丁胺溶于有机溶剂,加热回流反应,减压浓缩,得到中间体v,反应方程式如下:
中间体iv中间体v;
在步骤s6中,将中间体v溶于有机溶剂,加入第二钯催化剂,于氢气氛围下室温搅拌反应,得到消旋沙丁胺醇,反应方程式如下:
中间体v消旋沙丁胺醇。
优选地,在步骤s1中,5-溴水杨醛与硼氢化钠的摩尔比为1:(1.1-1.5),有机溶剂选自乙醇、甲醇、n,n-二甲基甲酰胺、四氢呋喃中的一种。
优选地,在步骤s2中,有机溶剂选自n,n-二甲基甲酰胺、四氢呋喃、甲苯中的一种,卤化苄选自氯化苄或溴化苄。
优选地,在步骤s3中,中间体ii、乙烯基三氟硼酸钾、第一钯催化剂、碱的摩尔比为1:(1.0-1.5):(0.02~0.05):(2.0~4.0);
优选地,在步骤s3中,第一钯催化剂为1,1'-双二苯基膦二茂铁二氯化钯、四三苯基膦钯、醋酸钯和三苯基膦、氯化钯和三苯基膦中的一种;碱为三乙胺、n,n-二异丙基乙胺、碳酸铯、碳酸钾、磷酸钾中的一种,溶剂为异丙醇、正丙醇、四氢呋喃水溶液、甲苯中的一种。
优选地,在步骤s4中,中间体iii与n-溴代琥珀酰亚胺的摩尔比为1:(2-5)。
优选地,在步骤s5中,有机溶剂选自异丙醇、正丙醇、叔丁醇、正丁醇中的一种。
优选地,在步骤s6中,有机溶剂选自四氢呋喃、甲醇、乙醇中的一种,第二钯催化剂为钯碳催化剂,中间体v与第二钯催化剂的质量比为1:(0.05-0.2)。
优选地,第一钯催化剂为醋酸钯和三苯基膦时,醋酸钯和三苯基膦的摩尔比为1:3;第一钯催化剂为氯化钯和三苯基膦时,氯化钯和三苯基膦的摩尔比为1:3;四氢呋喃水溶液中四氢呋喃与水的体积比为9:1;第一钯催化剂为醋酸钯和三苯基膦时,将摩尔比为1:3的醋酸钯、三苯基膦一同加入反应体系中;第一钯催化剂为氯化钯和三苯基膦时,将摩尔比为1:3的氯化钯、三苯基膦一同加入反应体系中。
与现有技术相比,本发明所提供的一种消旋沙丁胺醇的制备方法,
1、首次采用价廉易得的5-溴水杨醛为反应原料合成沙丁胺醇,降低生产成本;并且,通过溴原子的引入,可使反应在5位上定向引入官能团,提高了反应的选择性和收率;
2、使用卤化苄对中间体i的羟基进行保护,有效避免了后续工艺过程中副反应的发生,杂质的引入;在脱保护时使用钯碳/h2体系代替剧毒的肼类化合物,大大降低了反应过程中的污染,符合“绿色化学”理念;
3、首次将suzuki-miyaura交叉偶联反应用于沙丁胺醇的合成中,使用催化量的钯催化剂,便可实现芳环上双键的高效引入,提高反应收率的同时降低了成本;
4、整个消旋沙丁胺醇的制备过程中不涉及高危剧毒试剂,反应条件温和、设备要求低,操作简单,可控性强,后处理时无需柱层析,环境污染小,既适合工业化生产又符合“绿色化学”理念。
附图说明
图1为中间体ii的1hnmr图;
图2为中间体ii的13cnmr图;
图3为中间体ii的esi-hrms图;
图4为中间体iii的1hnmr图;
图5为中间体iii的13cnmr图;
图6为中间体iii的esi-hrms图;
图7为中间体v的1hnmr图;
图8为中间体v的13cnmr图;
图9为中间体v的esi-hrms图;
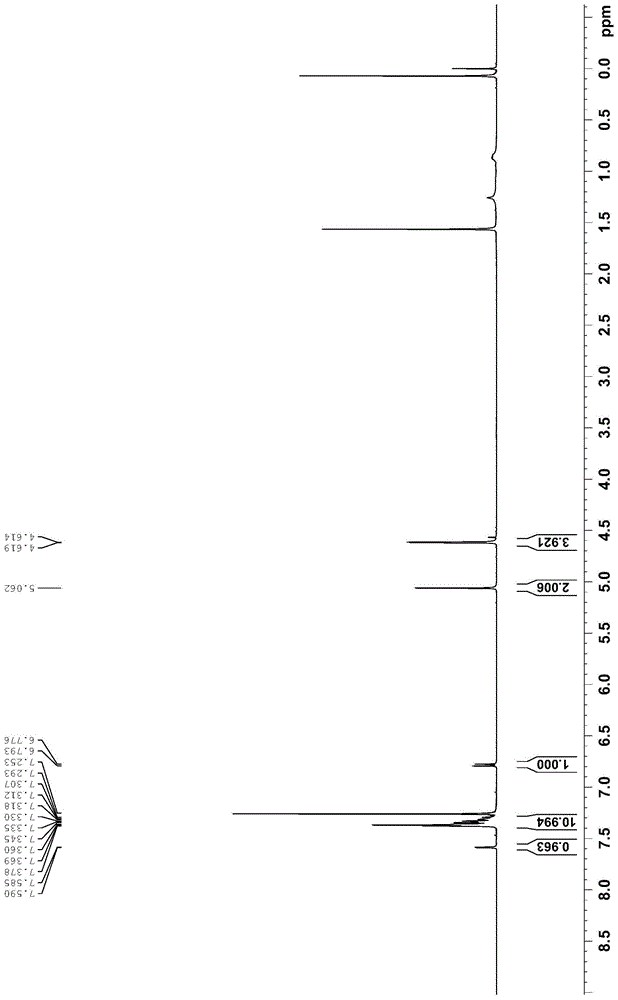
图10为消旋沙丁胺醇的1hnmr图;
图11为消旋沙丁胺醇的13cnmr图;
图12为消旋沙丁胺醇的esi-hrms图。
具体实施方式
为了使本领域技术人员能够更好的理解本发明,下面结合具体实施方式对本发明作进一步的详细说明。
实施例1
1)在1l三口瓶中将5-溴水杨醛(40.2g,200mmol)溶于无水乙醇(400ml),体系降温至0-5℃,加入硼氢化钠(9.1g,240mmol),加毕,搅拌2小时,停止反应,减压浓缩,残留物加入1n盐酸(400ml)、乙酸乙酯(200ml)萃取分液,萃取得到的有机相用水(400ml)洗涤,用无水硫酸钠干燥,抽滤,滤液减压浓缩,干燥得中间体i39.8g,收率98%,白色固体。
反应方程式为:
中间体i。
2)500ml三口瓶中,将中间体i(30.45g,150mmol)溶于n,n-二甲基甲酰胺(250ml),体系降温至0-5℃,在搅拌下加入氢化钠(60%,分散于液状石蜡)(12g,300mmol),加毕,搅拌10分钟后,向其中滴加溴化苄(51.3g,300mmol)。加毕,反应体系自然升至室温,搅拌6小时,停止反应,体系倒入冰水中,加入乙酸乙酯(200ml)萃取分液,萃取得到的有机相分别用水(200ml)、饱和食盐水(200ml)洗涤,无水硫酸钠干燥,抽滤,滤液减压浓缩,干燥得中间体ii56.5g,收率97%,白色固体。中间体ii的1hnmr、13cnmr和esi-hrms分别如图1-3所示,1hnmr(500mhz,cdcl3):δ7.58(d,j=2.3hz,1h),7.38-7.29(m,11h),6.78(d,j=8.7hz,1h),5.06(s,2h),4.61(d,j=2.6hz,4h);13cnmr(125mhz,cdcl3):δ154.6,137.7,136.1,131.0,130.5,129.1,128.1,127.9,127.5,127.2,127.1,126.7,112.9,112.8,72.3,69.8,66.1.hrms(esi):calcdforc21h19o2brna[m+na]+:407.0446,found407.0442.
反应方程式为:
中间体i中间体ii。
3)在500ml三口瓶中,将中间体ii(38.3g,100mmol)溶于四氢呋喃水溶液(200ml,其中四氢呋喃与水的体积比为9:1),加入乙烯基三氟硼酸钾(16.08g,120mmol)、氯化钯(0.36g,2mmol)、三苯基膦(1.57g,6mmol)、碳酸铯(65.2g,200mmol),体系回流反应12小时,停止反应,体系自然冷却至室温,加入水(150ml)、乙酸乙酯(150ml)萃取分液,萃取得到的有机相用饱和食盐水(100ml)洗涤,无水硫酸钠干燥,抽滤,滤液减压浓缩,将残留物加入乙酸乙酯和正己烷的混合液(100ml,其中乙酸乙酯和正己烷的体积比为1:9),打浆1小时,抽滤,滤饼用正己烷(50ml)洗涤,干燥得中间体iii30.1g,收率91%,白色固体。中间体iii的1hnmr、13cnmr和esi-hrms分别如图4-6所示,1hnmr(500mhz,cdcl3):δ7.54(s,1h),7.41-7.28(m,11h),6.88(d,j=8.4hz,1h),6.68(q,j=10.9hz,1h),5.64(d,j=17.6hz,1h),5.14(d,j=10.9hz,1h),5.11(s,2h),4.67(s,2h),4.63(s,2h);13cnmr(125mhz,cdcl3):δ155.6,138.0,136.6,135.8,130.1,128.1,127.9,127.4,127.3,127.0,126.7,126.4,126.1,111.4,111.2,72.1,69.6,66.7.hrms(esi):calcdforc23h22o2na[m+na]+:353.1517,found353.1516.
反应方程式为:
中间体ii中间体iii。
4)在500ml三口瓶中加入中间体iii(19.8g,60mmol)、二甲基亚砜(150ml)和水(1.5ml),搅拌10分钟后,加入n-溴代琥珀酰亚胺(21.4g,120mmol),室温反应3小时。停止反应,加入水(150ml)、乙酸乙酯(150ml),萃取分液,萃取得到的有机相用饱和食盐水(150ml)洗涤,无水硫酸钠干燥,抽滤,滤液减压浓缩,得中间体iv23.8g,收率93%,淡黄色油状物。
反应方程式为:
中间体iii中间体iv。
5)在500ml三口瓶中,将中间体iv(17.1g,40mmol)、叔丁胺(100ml)溶于异丙醇(100ml),加热回流6小时,停止反应,体系减压浓缩,残留物加入二氯甲烷和正己烷的混合液(70ml,其中二氯甲烷和正己烷的体积比为1:9),打浆1小时,抽滤,滤饼用正己烷(30ml)洗涤,干燥得中间体v13.6g,收率81%,白色固体。中间体v的1hnmr、13cnmr和esi-hrms分别如图7-9所示,1hnmr(500mhz,cdcl3):δ7.38-7.19(m,12h),6.85(d,j=8.4hz,1h),5.03(s,2h),4.63-4.58(m,2h),4.55(s,2h),4.52-4.50(m,1h),2.81-2.78(m,1h),2.56-2.52(m,1h),1.04(s,9h);13cnmr(125mhz,cdcl3):δ155.2,138.0,136.7,134.9,128.0,127.9,127.3,127.0,126.7,126.5,126.4,125.6,111.2,72.2,71.6,69.6,66.9,49.8,29.2,28.7.hrms(esi):calcdforc27h34no3[m+h]+:420.2539,found420.2537.
反应方程式为:
中间体iv中间体v。
6)在500ml三口瓶中,将中间体v(8.4g,20mmol)溶于四氢呋喃(200ml),加入钯碳催化剂(0.84g,钯碳催化剂中钯含量为10wt%),于氢气气氛中室温下快速搅拌12小时。停止反应,过滤,滤饼用四氢呋喃(10ml)洗涤,滤液减压浓缩,将残留物加入乙酸乙酯和正己烷的混合液(50ml,其中乙酸乙酯和正己烷的体积比为1:9),打浆1小时,抽滤,滤饼用正己烷(20ml)洗涤,干燥得消旋沙丁胺醇4.1g,收率86%,白色固体。消旋沙丁胺醇的1hnmr、13cnmr和esi-hrms分别如图10-12所示,1hnmr(400mhz,dmso-d6):δ7.25(d,j=1.5hz,1h),7.00(dd,j=8.2hz,2.0hz,1h),6.68(d,j=8.1hz,1h),4.47(s,2h),4.40(t,j=6.0hz,1h),2.52-2.49(m,2h),1.00(s,9h);13cnmr(100mhz,dmso-d6):δ153.1,134.6,127.9,125.0,124.8,114.0,72.4,58.3,50.8,49.5,28.9.hrms(esi):calcdforc13h22no3[m+h]+:240.1600,found240.1600.
反应方程式为:
中间体v消旋沙丁胺醇。
实施例2
1)在1l三口瓶中将5-溴水杨醛(40.2g,200mmol)溶于甲醇(400ml),体系降温至0-5℃,加入硼氢化钠(8.3g,220mmol),加毕,搅拌2小时,停止反应,减压浓缩,残留物加入1n盐酸(400ml)、乙酸乙酯(200ml)萃取分液,萃取得到的有机相用水(400ml)洗涤,用无水硫酸钠干燥,抽滤,滤液减压浓缩,干燥得中间体i38.6g,收率95%,白色固体。
2)500ml三口瓶中,将中间体i(30.45g,150mmol)溶于甲苯(250ml),体系降温至0-5℃,在搅拌下加入氢化钠(60%,分散于液状石蜡)(12g,300mmol),加毕,搅拌10分钟后,向其中滴加溴化苄(51.3g,300mmol)。加毕,反应体系自然升至室温,搅拌6小时,停止反应,体系倒入冰水中,加入乙酸乙酯(200ml)萃取,萃取得到的有机相分别用水(200ml)、饱和食盐水(200ml)洗涤,无水硫酸钠干燥,抽滤,滤液减压浓缩,干燥得中间体ii54.2g,收率93%,白色固体。
3)在500ml三口瓶中,将中间体ii(38.3g,100mmol)溶于正丙醇(200ml),加入乙烯基三氟硼酸钾(13.4g,100mmol)、醋酸钯(0.68g,3mmol)、三苯基膦(2.36g,9mmol)、n,n-二异丙基乙胺(51.8g,400mmol),体系回流反应12小时,停止反应,体系自然冷却至室温,加入水(150ml)、乙酸乙酯(150ml)萃取分液,萃取得到的有机相用饱和食盐水(100ml)洗涤,无水硫酸钠干燥,抽滤,滤液减压浓缩,将残留物加入乙酸乙酯和正己烷的混合液(100ml,其中乙酸乙酯和正己烷的体积比为1:9),打浆1小时,抽滤,滤饼用正己烷(50ml)洗涤,干燥得中间体iii24.8g,收率75%,白色固体。
4)在500ml三口瓶中加入中间体iii(19.8g,60mmol)、二甲基亚砜(150ml)和水(1.5ml),搅拌10分钟后,加入n-溴代琥珀酰亚胺(37.4g,210mmol),室温反应3小时。停止反应,加入水(150ml)、乙酸乙酯(150ml),萃取分液,萃取得到的有机相用饱和食盐水(150ml)洗涤,无水硫酸钠干燥,抽滤,滤液减压浓缩,得中间体iv24.1g,收率94%,淡黄色油状物。
5)在500ml三口瓶中,将中间体iv(17.1g,40mmol)、叔丁胺(100ml)溶于正丙醇(100ml),加热回流6小时,停止反应,体系减压浓缩,残留物加入二氯甲烷和正己烷的混合液(70ml,其中二氯甲烷和正己烷的体积比为1:9),打浆1小时,抽滤,滤饼用正己烷(30ml)洗涤,干燥得中间体v12.6g,收率75%,白色固体。
6)在500ml三口瓶中,将中间体v(8.4g,20mmol)溶于甲醇(200ml),加入钯碳催化剂(0.42g,钯碳催化剂中钯含量为10wt%),于氢气气氛中室温下快速搅拌24小时。停止反应,过滤,滤饼用四氢呋喃(10ml)洗涤,滤液减压浓缩,将残留物加入乙酸乙酯和正己烷的混合液(50ml,其中乙酸乙酯和正己烷的体积比为1:9),打浆1小时,抽滤,滤饼用正己烷(20ml)洗涤,干燥得消旋沙丁胺醇4.0g,收率81%,白色固体。
实施例3
1)在1l三口瓶中将5-溴水杨醛(40.2g,200mmol)溶于n,n-二甲基甲酰胺(400ml),体系降温至0-5℃,加入硼氢化钠(11.4g,300mmol),加毕,搅拌2小时,停止反应,减压浓缩,残留物加入1n盐酸(400ml)、乙酸乙酯(200ml)萃取分液,萃取得到的有机相用水(400ml)洗涤,用无水硫酸钠干燥,抽滤,滤液减压浓缩,干燥得中间体i39.8g,收率98%,白色固体。
2)500ml三口瓶中,将中间体i(30.45g,150mmol)溶于四氢呋喃(250ml),体系降温至0-5℃,在搅拌下加入氢化钠(60%,分散于液状石蜡)(12g,300mmol),加毕,搅拌10分钟后,向其中滴加溴化苄(51.3g,300mmol)。加毕,反应体系自然升至室温,搅拌6小时,停止反应,体系倒入冰水中,加入乙酸乙酯(200ml)萃取,萃取得到的有机相分别用水(200ml)、饱和食盐水(200ml)洗涤,无水硫酸钠干燥,抽滤,滤液减压浓缩,干燥得中间体ii55.3g,收率95%,白色固体。
3)在500ml三口瓶中,将中间体ii(38.3g,100mmol)溶于异丙醇(200ml),加入乙烯基三氟硼酸钾(20.1g,150mmol)、1,1'-双二苯基膦二茂铁二氯化钯(3.65g,5mmol)、三乙胺(30.3g,300mmol),体系回流反应12小时,停止反应,体系自然冷却至室温,加入水(150ml)、乙酸乙酯(150ml)萃取分液,萃取得到的有机相用饱和食盐水(100ml)洗涤,无水硫酸钠干燥,抽滤,滤液减压浓缩,将残留物加入乙酸乙酯和正己烷的混合液(100ml,其中乙酸乙酯和正己烷的体积比为1:9),打浆1小时,抽滤,滤饼用正己烷(50ml)洗涤,干燥得中间体iii38.1g,收率85%,白色固体。
4)在500ml三口瓶中加入中间体iii(19.8g,60mmol)、二甲基亚砜(150ml)和水(1.5ml),搅拌10分钟后,加入n-溴代琥珀酰亚胺(53.4g,300mmol),室温反应3小时。停止反应,加入水(150ml)、乙酸乙酯(150ml),萃取分液,萃取得到的有机相用饱和食盐水(150ml)洗涤,无水硫酸钠干燥,抽滤,滤液减压浓缩,得中间体iv22.5g,收率88%,淡黄色油状物。
5)在500ml三口瓶中,将中间体iv(17.1g,40mmol)、叔丁胺(100ml)溶于叔丁醇(100ml),加热回流6小时,停止反应,体系减压浓缩,残留物加入二氯甲烷和正己烷的混合液(70ml,其中二氯甲烷和正己烷的体积比为1:9),打浆1小时,抽滤,滤饼用正己烷(30ml)洗涤,干燥得中间体v13.6g,收率78%,白色固体。
6)在500ml三口瓶中,将中间体v(8.4g,20mmol)溶于乙醇(200ml),加入钯碳催化剂(1.68g,钯碳催化剂中钯含量为10wt%),于氢气气氛中室温下快速搅拌4小时。停止反应,过滤,滤饼用四氢呋喃(10ml)洗涤,滤液减压浓缩,将残留物加入乙酸乙酯和正己烷的混合液(50ml,其中乙酸乙酯和正己烷的体积比为1:9),打浆1小时,抽滤,滤饼用正己烷(20ml)洗涤,干燥得消旋沙丁胺醇4.2g,收率88%,白色固体。
实施例4
1)在1l三口瓶中将5-溴水杨醛(40.2g,200mmol)溶于四氢呋喃(400ml),体系降温至0-5℃,加入硼氢化钠(9.1g,240mmol),加毕,搅拌2小时,停止反应,减压浓缩,残留物加入1n盐酸(400ml)、乙酸乙酯(200ml)萃取分液,萃取得到的有机相用水(400ml)洗涤,用无水硫酸钠干燥,抽滤,滤液减压浓缩,干燥得中间体i38.2g,收率94%,白色固体。
2)500ml三口瓶中,将中间体i(30.45g,150mmol)溶于n,n-二甲基甲酰胺(250ml),体系降温至0-5℃,在搅拌下加入氢化钠(60%,分散于液状石蜡)(12g,300mmol),加毕,搅拌10分钟后,向其中滴加氯化苄(38.0g,300mmol)。加毕,反应体系自然升至室温,搅拌6小时,停止反应,体系倒入冰水中,加入乙酸乙酯(200ml)萃取,萃取得到的有机相分别用水(200ml)、饱和食盐水(200ml)洗涤,无水硫酸钠干燥,抽滤,滤液减压浓缩,干燥得中间体ii55.9g,收率96%,白色固体。
3)在500ml三口瓶中,将中间体ii(38.3g,100mmol)溶于甲苯(200ml),加入乙烯基三氟硼酸钾(16.08g,120mmol)、四三苯基膦钯(2.31g,2mmol)、碳酸钾(27.6g,200mmol),体系回流反应12小时,停止反应,体系自然冷却至室温,加入水(150ml)、乙酸乙酯(150ml)萃取分液,萃取得到的有机相用饱和食盐水(100ml)洗涤,无水硫酸钠干燥,抽滤,滤液减压浓缩,将残留物加入乙酸乙酯和正己烷的混合液(100ml,其中乙酸乙酯和正己烷的体积比为1:9),打浆1小时,抽滤,滤饼用正己烷(50ml)洗涤,干燥得中间体iii27.1g,收率82%,白色固体。
4)在500ml三口瓶中加入中间体iii(19.8g,60mmol)、二甲基亚砜(150ml)和水(1.5ml),搅拌10分钟后,加入n-溴代琥珀酰亚胺(21.4g,120mmol),室温反应3小时。停止反应,加入水(150ml)、乙酸乙酯(150ml),萃取分液,萃取得到的有机相用饱和食盐水(150ml)洗涤,无水硫酸钠干燥,抽滤,滤液减压浓缩,得中间体iv23.8g,收率93%,淡黄色油状物。
5)在500ml三口瓶中,将中间体iv(17.1g,40mmol)、叔丁胺(100ml)溶于正丁醇(100ml),加热回流6小时,停止反应,体系减压浓缩,残留物加入二氯甲烷和正己烷的混合液(70ml,其中二氯甲烷和正己烷的体积比为1:9),打浆1小时,抽滤,滤饼用正己烷(30ml)洗涤,干燥得中间体v11.9g,收率71%,白色固体。
6)在500ml三口瓶中,将中间体v(8.4g,20mmol)溶于乙醇(200ml),加入钯碳催化剂(0.84g,钯碳催化剂中钯含量为10wt%),于氢气气氛中室温下快速搅拌12小时。停止反应,过滤,滤饼用四氢呋喃(10ml)洗涤,滤液减压浓缩,将残留物加入乙酸乙酯和正己烷的混合液(50ml,其中乙酸乙酯和正己烷的体积比为1:9),打浆1小时,抽滤,滤饼用正己烷(20ml)洗涤,干燥得消旋沙丁胺醇4.1g,收率86%,白色固体。
以上对本发明所提供的一种消旋沙丁胺醇的制备方法进行了详细的介绍。本文中应用了具体个例对本发明的原理及实施方式进行了阐述,以上实施例的说明只是用于帮助理解本发明的方法和中心思想,所提到的方向用语,例如:上、下、左、右、前或后等,仅是参考附图的方向,使用的方向用语是用来说明并非用来限制本发明。应当指出,对于本技术领域的一般技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明权利要求的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!