同时分离克伦特罗、沙丁胺醇和莱克多巴胺的分子印迹柱的制作方法
本发明涉及一种分子印迹固相萃取柱,特别涉及能同时分离动物产品和饲料中克伦特罗、沙丁胺醇和莱克多巴胺的三合一分子印迹固相萃取柱。
背景技术:
:
“瘦肉精”是β2-受体激动剂类药物。利用分子印迹技术对瘦肉精进行检测,是将待检的瘦肉精化合物作为模板分子,采用功能单体分子与模板分子结合,交联剂和引发剂促使单体之间进行交联,从而得到针对特定模板分子的印迹化合物。目前,克伦特罗、莱克多巴胺和沙丁胺醇是违法滥用较严重的三种“瘦肉精”化合物。
目前用于检测瘦肉精的分子印迹柱多为每次只能检测一种瘦肉精化合物,没有有效解决样本中违法滥用较严重的三种瘦肉精分子的同时分离富集问题。如姜久英等CN 102955012A用甲基丙烯酸作为单体分子,制备了克伦特罗的分子印迹整体柱。李建平等CN104945552A同样用甲基丙烯酸作为单体分子,合成了克伦特罗的分子印迹聚合物。这些专利只描述了克伦特罗分子的分子印迹材料的一种合成方法,还未涉及样本中的其他种类瘦肉精分子的净化。汤秩伟等CN104535759A描述了一种克伦特罗的仿生免疫柱,利用分子印迹材料装填,并且在柱中完成现场检测。在此发明中,克伦特罗的分子印迹材料作为一个样本中克伦特罗分子的捕获物,直接捕获待检样本中的克伦特罗分子并直接显色检测,不涉及到克伦特罗分子的分离纯化。
技术实现要素:
本发明的目的是提供一种分子印迹固相萃取柱,可以同时从待检样本中分离并纯化克伦特罗、沙丁胺醇以及莱克多巴胺,用于单独或者同时分离分析饲料、动物尿液以及动物组织中的这三种最常见的瘦肉精类化合物。
本发明提供一种能同时分离克伦特罗、沙丁胺醇和莱克多巴胺的三合一分子印迹柱,其特征在于,所述分子印迹柱中具有能分别识别分离克伦特罗、沙丁胺醇和莱克多巴胺的三种分子印迹聚合物。
克伦特罗、沙丁胺醇和莱克多巴胺三种分子印迹聚合物在柱管中装填的比例不限,本领域技术人员可根据实际需要确定用量。
当不能确定检测样本中三种瘦肉精类化合物比例时,三种分子印迹聚合物的优选比例(摩尔比)为:
克伦特罗聚合物:沙丁胺醇聚合物:莱克多巴胺聚合物=1:1~1.5:1~1.5。
沙丁胺醇聚合物:莱克多巴胺聚合物用量可以较大的原因是:单一使用沙丁胺醇分子印迹聚合物也可以对克伦特罗有一定比例的富集,莱克多巴胺分子印迹聚合物只能对莱克多巴胺实现吸附富集。
本发明研究筛选制备所述三种分子印迹聚合物最合适的功能单体和交联剂,通过优化具体目标分析物与功能单体、交联剂的种类、用量比例,将优化结果确定如下:
表1本发明发现的最适合的功能单体和交联剂
三种分子印迹聚合物都是用偶氮二异丁腈作为引发剂。
实验中发现,不合适的功能单体和交联剂在制备聚合物时会导致聚合物微球粒径不均一,易凝结成块,进行装柱后吸附富集效果不理想。
本发明还发现,聚合物的微球直径对分离效果也有影响,不合适的粒径会导致装柱效果不理想,粒径过大或过小,会影响装柱后进行吸附试验时溶剂通过分子印迹柱的流速,从而导致吸附富集效果。本发明通过实验找到了三种分子印迹聚合物可用的微球直径是10-1005μm,优选的微球直径是20-60μm。
本发明的分子印迹柱中具有筛板,筛板的数量是2~4个。筛板的孔径在5μm到20μm之间。优选的筛板孔径是8-10μm。筛板的材质选自聚乙烯、高密度聚乙烯及聚丙烯,优选聚丙烯。
本发明还发现,不合适的筛板孔径会影响装柱后进行吸附试验时溶剂通过分子印迹柱的流速,从而导致吸附富集效果。
柱管材质选自聚丙烯、聚苯乙烯及PEEK材质,优选聚丙烯。
本发明的三合一分子印迹柱能够同时分离并纯化待测样品中克伦特罗、沙丁胺醇和莱克多巴胺三种物质。所述样品包括组织,尿液和饲料等。富集分离出的克伦特罗、沙丁胺醇和莱克多巴胺可以直接用于高效液相色谱-串联质谱或其它方法检测。
本发明的三合一分子印迹柱的制备方法如下:
1、三种分子印迹聚合物的制备方法
包括功能单体分子和模板分子的混合及溶解;加入引发剂和交联剂;催化反应;制备分子印迹微球;模板洗脱这五部分。
其特征是:所述制备分子印迹微球步骤中,在水相体系中进行悬浮聚合,使模板分子、功能单体、交联剂等保持良好的分散,利用悬浮聚合法得到的分子印迹聚合物粒径均一,无需研磨过筛就可直接装柱使用。
例如,在本发明的一个实例中,使用4%的聚乙烯醇水溶液(4g聚乙烯醇溶解于100mL水)作为溶剂。
而现有技术的制备方法是沉淀聚合,即,采用的溶剂通常是非极性的有机溶剂,如乙腈、氯仿等非极性溶剂。其缺点是得到的聚合物不能直接应用,需要再经研磨过筛工序,使粒径均一后才能使用。而且,这些非极性溶剂容易造成聚合过程中的环境污染。
2、三种分子印迹聚合物微球的装填方式
可以将克伦特罗、沙丁胺醇和莱克多巴胺三种聚合物微球分层装填,且三种聚合物微球均可位于三合一分子印迹柱的任一层(见图3);采用此种装填方式,每两层间用筛板分隔开。
也可以将三种聚合物微球先混合后,不分层一起装填在柱中;所述混合,可以是均匀混合,也可以是随机混合,都可以达到本发明的效果。
以三种分子印迹聚合物作为填料按照一定方式混合或直接分层装填后得到混合分子印迹固相萃取柱,优化了使用此分子印迹柱进行三种“瘦肉精”类药物分离富集过程中的活化、上样、淋洗、洗脱过程,确定了每步所需溶剂种类及用量(详见具体实施方式)。
使用此分子印迹固相萃取柱从不同样本中分离净化克伦特罗、沙丁胺醇和莱克多巴胺,在经过固相萃取过程后,将洗脱液经过氮气吹干以合适的溶剂复溶后上机测定,回收率可达80%-95%。适合作为分析前处理使用。
本发明的有益效果是:
1、首次将三种瘦肉精化合物克伦特罗、沙丁胺醇和莱克多巴胺制备的分子印迹化合物装填在同一根分子印迹固相萃取柱中,实现了样本中三种瘦肉精分子的同时分离;
2、通过实验,选择了合适的功能单体和交联剂,经实验证明,制备得到的三种分子印迹聚合物分离富集效果好;
3、采用了水相环境中悬浮聚合法制备三种分子印迹聚合物微球,避免了常规的本体聚合法需要磨碎、过筛等过程。制备得到的微球粒径均一,适合作为填料。
4、分子印迹固相萃取柱经简单溶剂处理后可反复使用,重复使用次数可达10次,经济环保。
附图说明
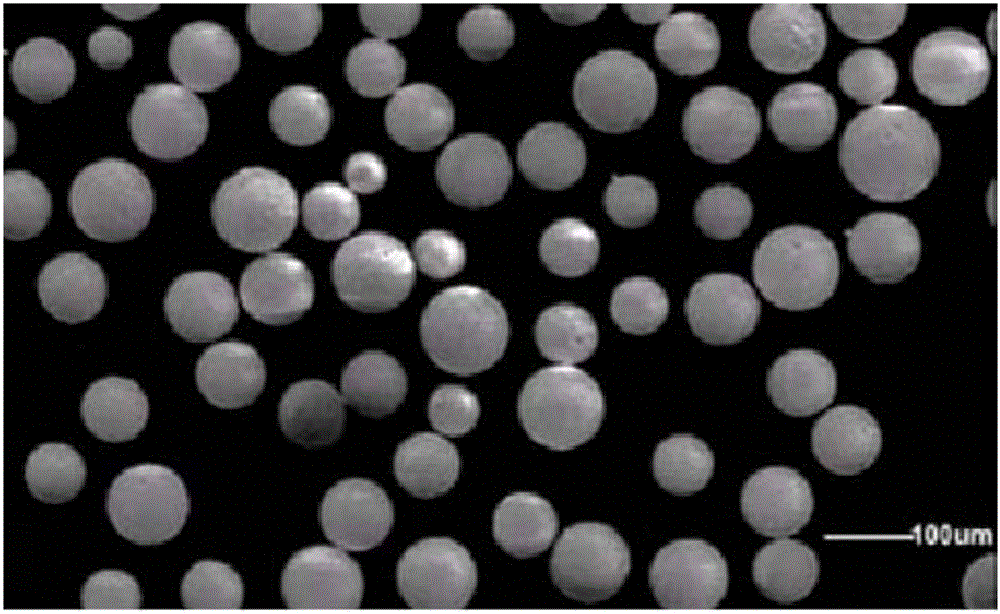
图1:克伦特罗分子印迹聚合物微球扫描电镜结构图;
图2:沙丁胺醇分子印迹聚合物微球扫描电镜结构图;
图3:莱克多巴胺分子印迹聚合物微球扫描电镜结构图;
图4:克伦特罗、沙丁胺醇和莱克多巴胺三合一分子印迹柱模式图,
左图为三种聚合物微球分层装填的三合一分子印迹柱模式图,其中1是柱管,2是筛板,3、4、5分别是三种瘦肉精化合物(沙丁胺醇、莱克多巴胺、克伦特罗)的分子印迹聚合物填料,6是上堵头,7是下堵头;
右图为三种聚合物微球混合装填的三合一分子印迹柱模式图,其中1是柱管,2是筛板,6是上堵头,7是下堵头,8是三种瘦肉精化合物的分子印迹聚合物混合物;
图5:克伦特罗、沙丁胺醇和莱克多巴胺标准品经三合一分子印迹柱净化后的高效液相色谱检测图谱;
图6:猪尿液中克伦特罗气相色谱-质谱检测图谱;
图7:饲料样本中沙丁胺醇高效液相检测图谱;
图8:组织样本中莱克多巴胺的高效液相色谱-串联质谱检测图谱。
图9:以TRIM做交联剂时得到的沙丁胺醇分子印迹聚合物
图10:不合适功能单体得到的克伦特罗分子印迹聚合物
图11:不合适比例的模版分子、功能单体和交联剂反应条件下得到的沙丁胺醇分子印迹聚合物
图12:200mL 4%的聚乙烯醇1788(PVA 1788)做溶剂时得到的沙丁胺醇分子印迹聚合物
具体实施方式
以下实施例仅用于举例说明本发明的方法和装置,并不限定本发明的范围。实施例中出现的化合物代号的中英文名称如下表所示。
中英文对照及缩写表
实施例1克伦特罗分子印迹聚合物的制备
1.称取2g克伦特罗加入到20mL水中,加入2mL0.1moL/L的盐酸使其充分溶解;用1moL/L的NaOH调pH值至12以上;用等体积的氯仿萃取三次,萃取液经3g无水硫酸钠脱水,50℃旋转真空干燥,得白色或近乎白色的克伦特罗结晶性粉末。
2.将100mL 4%的聚乙烯醇1788加入四口瓶中,400r/min搅拌同时通氮气10min。
3.在另一容器中,将已除去阻聚剂的0.8mL DEAEM、5.0mL EGDMA和10.0mL辛醇、0.5g克伦特罗,超声混匀20min,然后加入120mg AIBN、5mL氯仿继续超声5min,超声时加入冰块,保持低温。保持搅拌速度,保证良好的单体分散。
4.持续通氮气,70℃,反应24h。
5.聚合反应结束后将得到的聚合物微球滤出,用水冲洗三次后倒入200mL烧杯中,80℃去离子水搅拌60min。
6.依次用50mL甲醇洗涤三次、50mL丙酮洗涤两次,除去溶胀剂、残余有机单体等有机物。
7.用甲醇:乙酸(80:20,v/v)溶液索氏萃取法洗脱模板24h;同样方法用甲醇处理24h,除去模板分子。
8.50℃真空干燥聚合物微球6h至衡重,获得平均粒径大小为80μm、表面疏松多孔的小球,其电镜扫描图如图1所示。
实施例2沙丁胺醇分子印迹聚合物的制备
1.将100mL 4%的聚乙烯醇1788(PVA 1788)加入四口瓶中,400r/min搅拌同时通氮气10min。
2.在另一容器中,将已除去阻聚剂的0.8mL MAA、5.0mL EGDMA和10.0mL辛醇、0.3g沙丁胺醇,超声混匀20min,然后加入120mg AIBN、5mL氯仿继续超声5min,超声时加入冰块,保持低温。保持搅拌速度,保证良好的单体分散。
3.持续通氮气,70℃,反应24h。
4.聚合反应结束后将得到的聚合物微球滤出,用水冲洗三次后倒入200mL烧杯中,80℃去离子水搅拌60min。
5.依次用50mL甲醇洗涤三次、50mL丙酮洗涤两次,除去溶胀剂、残余有机单体等有机物。
6.用甲醇:乙酸(80:20,v/v)溶液索氏萃取法洗脱模板24h;同样方法用甲醇处理24h,除去模板分子。
7.50℃真空干燥聚合物微球6h至衡重,获得平均粒径大小为80μm、表面疏松多孔的小球,其电镜扫描图如图2所示。
实施例3莱克多巴胺分子印迹聚合物的制备
1.将100mL 4%的聚乙烯醇1788(PVA 1788)加入四口瓶中,400r/min搅拌同时通氮气10min。
2.在另一容器中,将已除去阻聚剂的0.8mLMAA、5.0mL EGDMA和10.0mL辛醇、0.6g莱克多巴胺,超声混匀20min,然后加入120mg AIBN、5mL氯仿继续超声5min,超声时加入冰块,保持低温。保持搅拌速度,保证良好的单体分散。
3.持续通氮气,70℃,反应24h。
4.聚合反应结束后将得到的聚合物微球滤出,用水冲洗三次后倒入200mL烧杯中,80℃去离子水搅拌60min。
5.依次用50mL甲醇洗涤三次、50mL丙酮洗涤两次,除去溶胀剂、残余有机单体等有机物。
6.用甲醇:乙酸(80:20,v/v)溶液索氏萃取法洗脱模板24h;同样方法用甲醇处理24h,除去模板分子。
7.50℃真空干燥聚合物微球6h至衡重,获得平均粒径大小为80μm、表面疏松多孔的小球,其电镜扫描图如图3所示。
实施例4克伦特罗、沙丁胺醇和莱克多巴胺的三合一分子印迹柱制备
1.取分子印迹柱空柱管、筛板、上堵头和下堵头,用去离子水超声波清洗后,烘干。
2.称取制备好的克伦特罗、沙丁胺醇和莱克多巴胺分子印迹聚合物各100mg,分别用2mL甲醇:异丙醇(2:1,v:v)溶胀。
3.取分子印迹柱空柱管,装入下筛板。
4.吸取0.5mL溶胀好的克伦特罗分子印迹聚合物,湿法装入空柱管中,装入上筛板,至上筛板紧压住聚合物微球。
5.吸取0.5mL沙丁胺醇分子印迹聚合物,加入到装有克伦特罗聚合物的柱管上层,加入到第一层上筛板上方,再加入一层筛板,至紧压住沙丁胺醇分子印迹聚合物微球。
6.吸取0.5mL莱克多巴胺分子印迹聚合物,加入到柱管中,加在第二层上筛板上方,再加入一层上筛板,至紧压住莱克多巴胺分子印迹聚合物微球。
7.或混合三种分子印迹聚合物制备分子印迹柱,具体实施过程:分别吸取4,5,6中分子印迹聚合物,混合后加入到装有下筛板的空柱管中,装入上筛板,压紧聚合物微球。
8.取7.5mL甲醇:异丙醇(2:1,v:v)洗涤装好的分子印迹柱。
9.在洗涤完成的分子印迹柱中加入2mL甲醇:异丙醇(2:1,v:v),堵上上堵头与下堵头,4℃保存,其示意图如图4所示。
实施例5利用克伦特罗、沙丁胺醇和莱克多巴胺的三合一分子印迹柱分离回收克伦特罗、沙丁胺醇和莱克多巴胺
1.称取克伦特罗标准品10mg,加甲醇溶解后,定容到100mL;称取沙丁胺醇标准品各5mg,分别加甲醇溶解后,定容到100mL。称取莱克多巴胺标准品2.5mg,加入甲醇溶解后,定容到100mL。这三种溶液作为克伦特罗、沙丁胺醇和莱克多巴胺的母液。
2.取克伦特罗母液1mL,用甲醇稀释,定容到100mL;同样分别取沙丁胺醇和莱克多巴胺的母液各1mL,用甲醇稀释到100mL。得到这三种溶液的工作液。
3.各取上述3中标准品的工作溶液1mL,混合后作为三合一分子印迹柱的检测溶液。
4.克伦特罗、沙丁胺醇和莱克多巴胺的三合一分子印迹柱(后简称印迹柱)用10mL甲醇:乙酸(9:1,v/v)洗涤。
5.用2mL甲醇冲洗印迹柱,2mL磷酸溶液(3mM,pH 3.4):乙腈(3:7,v/v)活化印迹柱,使之不抽干。
6.取三合一分子印迹柱检测溶液3mL,加入到上述分子印迹柱后,抽干。
7.用20%乙腈水溶液(2:8,v/v)淋洗印迹柱,抽干。
8.用3×1mL甲醇:乙酸(9:1,v/v)洗脱。
9.洗脱产物于50℃下氮气吹干后用1mL甲醇复溶,而后使用高效液相色谱仪检测。
色谱条件:
C18反相色谱柱(4.6mm×250mm,5μm);
荧光检测器:激发波长为226nm,发射波长为305nm
流速:1mL.min-1;
柱温:40℃;
流动相:水溶液(0.087%戊烷磺酸钠和2%乙酸):乙腈=80:20(v/v),使用前需过膜脱气。
10.分子印迹柱分离的克伦特罗、沙丁胺醇和莱克多巴胺高效液相色谱检测图谱如图5所示。根据高效液相检测结果及上样量计算样品的上样回收率。回收结果显示,克伦特罗回收率102%;沙丁胺醇回收率96%;莱克多巴胺回收率95%。
实施例6利用克伦特罗、沙丁胺醇和莱克多巴胺的三合一分子印迹柱分离回收猪尿液中的克伦特罗
1.采用阴性样本加标的方法检测猪尿液中的克伦特罗。取阴性的猪尿液样本,加入一定量的克伦特罗标准品,用本发明的三合一分子印迹柱净化回收后,用气相色谱-质谱法检测结果。
2.称取20.00mg克伦特罗标准品(纯度>99.5%),精确至0.00001g,溶于甲醇并定容至100mL,该贮备液浓度为200mg/L。
3.用25mmoL/L(pH6.7)的乙酸铵缓冲液稀释猪尿样(1∶1,V/V),尿样以3000r/min离心10分钟。取上清,在20mL猪尿液样本中,加入200μL浓度为200mg/mL的克伦特罗储备液。制备猪尿样本检测液。
4.克伦特罗、沙丁胺醇和莱克多巴胺的三合一分子印迹柱(后简称印迹柱)用10mL甲醇:乙酸(9:1,v/v)洗涤。
5.用2mL甲醇冲洗印迹柱,2mL磷酸溶液(3mM,pH 3.4):乙腈(3:7,v/v)活化印迹柱,使之不抽干。
6.取10mL处理好的猪尿样本检测液,加入到上述三合一分子印迹柱中。
用去离子水淋洗,抽干。
7.用20%乙腈水溶液(2:8,v/v)淋洗印迹柱,抽干。
8.用3mL甲醇:乙酸(9:1,v/v)洗脱。
9.取净化后的样品用氮气吹干,加入100μL BSTFA衍生化试剂,振荡,密封,置于75℃的烘箱中衍生30min,冷却,用氮气吹干。加入200μL甲苯溶解,振荡,上机分析。
气相色谱-质谱条件:
气相色谱-质谱仪(美国Thermo公司Trace DSQ),
仪器进样口温度为260℃;进样量为1.0μL,不分流模式:柱始温为70℃保持1min。
以25℃/min升至200℃,保持6min,再以25℃/min升至280℃,保持2min。EI源电子轰击能70eV,检测器温度200℃,接口温度250℃,质量扫描范围为70~400AMU,溶剂延迟8min。
10.气相色谱-质谱检测结果如图6所示。加标样本上样量为克伦特罗1mg/L,检测结果为0.94mg/L;回收率为94%。
实施例7利用克伦特罗、沙丁胺醇和莱克多巴胺的三合一分子印迹柱分离回收饲料样本中的沙丁胺醇
1.采用阴性样本加标的方法检测饲料样本中的沙丁胺醇。取阴性饲料样本,加入一定量的沙丁胺醇标准品,用本发明的三合一分子印迹柱净化回收后,用高效液相色谱法检测结果。
2.取20mg沙丁胺醇标准品,用甲醇溶解,并定容到100mL,配制200mg/L的沙丁胺醇标准品。
3.称取10g配合饲料样本,于250mL玻璃具塞三角瓶中,加入200mg/L的沙丁胺醇标准溶液100μL,静置20min,加入0.2%盐酸-甲醇(10:90,v/v)提取液100mL,振荡30min,于6000r/min速率下离心10min。
4.取上清液置于离心试管中,45℃下氮气吹干,用10mL水涡旋30s溶解残渣,加入2mL二氯甲烷涡旋30s,于5 000r/min速率下离心5min。取上层水相,置于另一离心管中,用氨水调节其pH至约7.0,待净化。
5.克伦特罗、沙丁胺醇和莱克多巴胺的三合一分子印迹柱(后简称印迹柱)用10mL甲醇:乙酸(9:1,v/v)洗涤。
6.用2mL甲醇冲洗印迹柱,2mL磷酸溶液(3mM,pH 3.4):乙腈(3:7,v/v)活化印迹柱,使之不抽干。
7.将上述配制的上样溶液加入到印迹柱后,抽干。
8.用20%乙腈水溶液(2:8,v/v)淋洗印迹柱,抽干。
9.用3×1mL甲醇:乙酸(9:1,v/v)洗脱。
洗脱产物于50℃下氮气吹干后用1mL甲醇复溶,而后使用高效液相色谱仪检测。
色谱条件如下:
色谱柱:AgiLentEcLipse XDB-C18柱(150mm×4.6mm,5μm);
流动相:4mmoL/L戊烷磺酸钠溶液(含0.2%甲酸)-乙腈(75:25,v/v);
流速:1mL/min;
进样量:10μL;
激发波长:λex=226nm、发射波长λem=305nm。
10.检测图谱如图7所示,根据检测结果计算回收率。饲料样本中的沙丁胺醇加标量为200ug/g,根据液相色谱计算得到的回收量为17.4ug/g,回收率为87%。
实施例8利用克伦特罗、沙丁胺醇和莱克多巴胺的三合一分子印迹柱分离回收组织样本中的莱克多巴胺
1.采用阴性样本加标的方法检测组织样本中的莱克多巴胺。取莱克多巴胺检测阴性的猪肝脏样本,加入一定量的莱克多巴胺标准品,用本发明的三合一分子印迹柱净化回收后,用高效液相色谱-质谱法检测结果。
2.样品提取:取20mg莱克多巴胺标准品,用甲醇溶解,并定容到100mL,配制200mg/L的莱克多巴胺标准品。称取5.00g猪肝脏组织匀浆组织于50mL离心管中,加入100uL浓度为200mg/L的莱克多巴胺标准品加入10mL丙酮,涡旋1min,超声1min,静置10min后,3000r/min离心10min。将上清液转入离心管中,残渣用10mL丙酮重复提取2次。合并3次提取液,3000r/min离心10min,将上清液转入鸡心瓶中,50℃下旋转蒸发至近干。
加入1mL乙酸铵溶液(25mmoL/L,pH 5.00±0.05)和20μLβ-葡萄糖苷酶,涡旋混匀,65℃静置2h。加入2mL硼酸钠缓冲液(25mmoL/L,pH10.3±0.1),涡动1min,再加入7mL乙酸乙酯,涡动1min,移至15mL离心管,3000r/min离心5min,取上清液,再重复萃取下层液体,合并上清。
3.克伦特罗、沙丁胺醇和莱克多巴胺的三合一分子印迹柱,用10mL甲醇:乙酸(9:1,v/v)洗涤。
4.用2mL甲醇冲洗印迹柱,2mL磷酸溶液(3mM,pH 3.4):乙腈(3:7,v/v)活化印迹柱,使之不抽干。
5.将上述提取的样本提取液加入到印迹柱后,抽干。
6.用20%乙腈水溶液(2:8,v/v)淋洗印迹柱,抽干。
7.用3×1mL甲醇:乙酸(9:1,v/v)洗脱。
8.洗脱产物于50℃下氮气吹干后用1mL甲醇复溶,而后使用高效液相色谱-串联质谱仪(LC-MS/MS)检测分析。
色谱条件:
色谱柱:BEH C18(100×2.1mm,1.7μm),柱温:30℃;
流动相A:0.1%甲酸和0.05%氨水溶液,B相:乙腈;
流速0.4mL/min;进样量5μL。
质谱条件:
离子源:电喷雾离子源;
扫描方式:正离子扫描;
检测方式:多反应监测;
电离电压3KV;
离子源温度150℃;
脱溶剂温度400℃;
碰撞气流速0.15mL/min;
锥孔气流速50L/h;
脱溶剂气流速550L/h。
10.莱克多巴胺LC-MS/MS检测结果如图8所示,根据检测结果计算回收率。根据检测结果,上样量为4ug/g,回收量为3.36ug/g;回收为率84%。
以下是对比实验
对比例1 不合适的交联剂导致分子印迹聚合物聚合效果不佳
1.将100mL 4%的聚乙烯醇1788(PVA 1788)加入四口瓶中,400r/min搅拌同时通氮气10min。
2.在另一容器中,将已除去阻聚剂的0.8mL MAA、5.0mL TRIM(三甲氧基丙基丙烯酸酯)和10.0mL辛醇、0.3g沙丁胺醇,超声混匀20min,然后加入120mg AIBN、5mL氯仿继续超声5min,超声时加入冰块,保持低温。保持搅拌速度,保证良好的单体分散。
3.持续通氮气,70℃,反应24h。
4.聚合反应结束后将得到的聚合物微球滤出,用水冲洗三次后倒入200mL烧杯中,80℃去离子水搅拌60min。
5.依次用50mL甲醇洗涤三次、50mL丙酮洗涤两次,除去溶胀剂、残余有机单体等有机物。
6.用甲醇:乙酸(80:20,v/v)溶液索氏萃取法洗脱模板24h;同样方法用甲醇处理24h,除去模板分子。
7.50℃真空干燥聚合物微球6h至衡重,得到的分子印迹聚合物电镜扫描如图9所示,聚合物凝聚成块,研磨后也不能得到粒径均一的聚合物颗粒,聚合效果不佳。
说明:在对比例1中也针对克伦特罗和莱克多巴胺进行了相同的交联剂选择试验尝试过程,其结果与沙丁胺醇分子印迹聚合物类似,分子印迹聚合物聚合效果不理想,克伦特罗和莱克多巴胺分子印迹聚合物凝聚成块,粒径不均匀。
对比例2 不合适的功能单体导致分子印迹聚合物聚合效果不佳
1.称取2g克伦特罗加入到20mL水中,加入2mL0.1moL/L的盐酸使其充分溶解;用1moL/L的NaOH调pH值至12以上;用等体积的氯仿萃取三次,萃取液经3g无水硫酸钠脱水,50℃旋转真空干燥,得白色或近乎白色的克伦特罗结晶性粉末。
2.将100mL 4%的聚乙烯醇1788加入四口瓶中,400r/min搅拌同时通氮气10min。
3.在另一容器中,将已除去阻聚剂的0.8mLMAA、5.0mL EGDMA和10.0mL辛醇、0.5g克伦特罗,超声混匀20min,然后加入120mg AIBN、5mL氯仿继续超声5min,超声时加入冰块,保持低温。保持搅拌速度,保证良好的单体分散。
4.持续通氮气,70℃,反应24h。
5.聚合反应结束后将得到的聚合物微球滤出,用水冲洗三次后倒入200mL烧杯中,80℃去离子水搅拌60min。
6.依次用50mL甲醇洗涤三次、50mL丙酮洗涤两次,除去溶胀剂、残余有机单体等有机物。
7.用甲醇:乙酸(80:20,v/v)溶液索氏萃取法洗脱模板24h;同样方法用甲醇处理24h,除去模板分子。
8.50℃真空干燥聚合物微球6h至衡重,得到的分子印迹聚合物如图10所示,聚合物凝结成块,研磨后也不能得到粒径均一的聚合物颗粒,聚合效果不佳。
说明:在对比例2中也针对沙丁胺醇和莱克多巴胺进行了功能单体(DEAEM)选择试验尝试过程,其结果与沙丁胺醇分子印迹聚合物类似,分子印迹聚合物聚合效果不理想,沙丁胺醇和莱克多巴胺分子印迹聚合物凝聚成块,粒径不均匀。
对比例3 不合适比例的模版分子、功能单体和交联剂导致的分子印迹聚合物聚合效果不佳
1.将100mL 4%的聚乙烯醇1788(PVA 1788)加入四口瓶中,400r/min搅拌同时通氮气10min。
2.在另一容器中,将已除去阻聚剂的1.6mL MAA、10.0mL EGDMA(或3.2mLMAA和20.0mLEGDMA)和10.0mL辛醇、0.3g沙丁胺醇,超声混匀20min,然后加入120mg AIBN、5mL氯仿继续超声5min,超声时加入冰块,保持低温。保持搅拌速度,保证良好的单体分散。
3.持续通氮气,70℃,反应24h。
4.聚合反应结束后将得到的聚合物微球滤出,用水冲洗三次后倒入200mL烧杯中,80℃去离子水搅拌60min。
5.依次用50mL甲醇洗涤三次、50mL丙酮洗涤两次,除去溶胀剂、残余有机单体等有机物。
6.用甲醇:乙酸(80:20,v/v)溶液索氏萃取法洗脱模板24h;同样方法用甲醇处理24h,除去模板分子。
7.50℃真空干燥聚合物微球6h至衡重,得到的分子印迹聚合物电镜图见图11,其聚合物表面不光滑,聚合效果不佳。
结果发现,分子印迹聚合物颗粒粒径不均一,呈现结块状态,在经研磨后也不能得到粒径均一的聚合物微粒,在装至空柱管后溶剂液体不能顺利流出。
说明:在对比例2中也同时针对克伦特罗和莱克多巴胺进行了模板分子、功能单体和交联剂的优化选择过程,其结果与沙丁胺醇分子印迹聚合物类似,不合适的模板分子、功能单体和交联剂的比例会导致分子印迹聚合物聚合效果不理想,克伦特罗和莱克多巴胺分子印迹聚合物凝聚成块,不能得到均匀的分子印迹聚合物微粒。
对比例4 聚合过程溶剂用量过大导致聚合物粒径偏小
1.将200mL 4%的聚乙烯醇1788(PVA 1788)加入四口瓶中,400r/min搅拌同时通氮气10min。
2.在另一容器中,将已除去阻聚剂的0.8mL MAA、5.0mL EGDMA和10.0mL辛醇、0.3g沙丁胺醇,超声混匀20min,然后加入120mg AIBN、5mL氯仿继续超声5min,超声时加入冰块,保持低温。保持搅拌速度,保证良好的单体分散。
3.持续通氮气,70℃,反应24h。
4.聚合反应结束后将得到的聚合物微球滤出,用水冲洗三次后倒入200mL烧杯中,80℃去离子水搅拌60min。
5.依次用50mL甲醇洗涤三次、50mL丙酮洗涤两次,除去溶胀剂、残余有机单体等有机物。
6.用甲醇:乙酸(80:20,v/v)溶液索氏萃取法洗脱模板24h;同样方法用甲醇处理24h,除去模板分子。
7.50℃真空干燥聚合物微球6h至衡重,得到的分子印迹聚合物电镜图见图12,分子印迹聚合物颗粒粒径大小均匀,但是粒径过小(小于5μm),在装入空柱管时容易出现渗漏现象,且溶剂流经速度过快,导致目标物分离效果不佳。
说明:在对比例3中也同时针对克伦特罗和莱克多巴胺分子印迹聚合物聚合过程中溶剂的用量进行了优化,其结果与沙丁胺醇类似,溶剂用量过大时导致分子印迹聚合物微粒粒径过小,不能很好地应用,分析结果不佳。
对比例5 分子印迹聚合物装柱量过多或过少导致分离效果不佳
1.取分子印迹柱空柱管、筛板、上堵头和下堵头,用去离子水超声波清洗后,烘干。称取2.制备好的克伦特罗、沙丁胺醇和莱克多巴胺分子印迹聚合物各50和200mg,分别用2mL甲醇:异丙醇(2:1,v:v)溶胀。
3.取分子印迹柱空柱管,装入下筛板。
4.吸取0.5mL溶胀好的克伦特罗分子印迹聚合物,湿法装入空柱管中,装入上筛板,至上筛板紧压住聚合物微球。
5.吸取0.5mL沙丁胺醇分子印迹聚合物,加入到装有克伦特罗聚合物的柱管上层,加入到第一层上筛板上方,再加入一层筛板,至紧压住沙丁胺醇分子印迹聚合物微球。
6.吸取0.5mL莱克多巴胺分子印迹聚合物,加入到柱管中,加在第二层上筛板上方,再加入一层上筛板,至紧压住莱克多巴胺分子印迹聚合物微球。
7.或混合三种分子印迹聚合物制备分子印迹柱,具体实施过程:分别吸取4,5,6中分子印迹聚合物,混合后加入到装有下筛板的空柱管中,装入上筛板,压紧聚合物微球。
8.取7.5mL甲醇:异丙醇(2:1,v:v)洗涤装好的分子印迹柱。
9.在洗涤完成的分子印迹柱中加入2mL甲醇:异丙醇(2:1,v:v),堵上上堵头与下堵头,4℃保存。
10.按照成功实施案例中实施案例8进行相同条件下的试验分析,结果发现:
1)每种分子印迹聚合物装柱量为50mg时,可能因为装柱量不够导致相同试验条件下,回收率效果偏低,根据高效液相检测结果及上样量计算样品的上样回收率。回收结果显示,克伦特罗回收率60%;沙丁胺醇回收率63%;莱克多巴胺回收率65%。不能满足分析方法要求;
2)每种分子印迹聚合物装柱量为100mg时,溶剂分离效果没有影响,但液体流经分子印迹聚合物的消耗时间长,也可能会增加杂质与目标分析物同时被洗脱的机率。
- 还没有人留言评论。精彩留言会获得点赞!