将氨再循环的制备α-羟基羧酸酯的方法与流程
将氨再循环的制备
α
‑
羟基羧酸酯的方法
1.本分案申请是基于申请号为201580048654.4、申请日为2015年9月4日、发明名称为“将氨再循环的制备α
‑
羟基羧酸酯的方法”的中国专利申请的分案申请。
技术领域
2.本发明涉及从氢氰酸出发制备α
‑
羟基羧酸酯(hce)的方法,其中将在相应的α
‑
羟基羧酰胺(hca)的醇解步骤中形成的氨在纯化步骤后再循环到氢氰酸制备工艺中。
背景技术:
3.hce的制备是从现有技术中充分已知的。更特别地,申请ep 2018362和wo 2013/026603描述了相应的方法,其中在压力下在液相中进行醇解,和在大于1巴的压力下蒸馏出形成的氨,或者通过气相移除形成的hce。
4.然而,采用这种现有技术的或甚至在其中于气相中进行所述醇解的已知方法中的重大问题是,所分离的氨,当被再循环到氢氰酸制备过程中时,导致即使在短时间后催化剂活性就显著丧失,和使得不可能经济地操作设备。
技术实现要素:
5.本发明要解决的问题因此是,后处理由从氢氰酸出发制备hce的过程中得到的氨,使得可以将其无任何问题地,即在反应时间、产率和品质方面没有损失的情况下,再循环到氢氰酸制备工艺中。
6.这些问题,和没有明确提及的其它问题,令人惊奇地由本发明提供从氢氰酸出发制备hce的方法解决,所述方法的特征在于,将在相应的α
‑
羟基羧酰胺的醇解步骤中形成的氨,在纯化步骤后,在仍包含至少一种烷基胺的情况下,再循环到氢氰酸制备工艺(hcn工艺)中。该解决方案是令人惊奇的,因为本发明的纯化步骤没有从由所述醇解反应形成的氨中移除所有的杂质,尤其是烷基胺,但hcn工艺可以无问题地运行。
7.在根据本发明的方法中可用的α
‑
羟基羧酰胺通常包括所有那些在所述羧酰胺基团的α位带有至少一个羟基基团的羧酰胺。
8.羧酰胺又是本领域中公知的。通常,其被理解为指具有式
‑
conr’r
”‑
的基团的化合物,其中r’和r”各自独立地是氢或具有1
‑
30个碳原子的基团,其尤其包含1
‑
20个,优选1
‑
10个和尤其是1
‑
5个碳原子。所述羧酰胺可带有1、2、3、4或更多个具有式
‑
conr’r”的基团。这些尤其包括式r(
‑
conr’r”)
n
的化合物,其中所述r基团是具有1
‑
30个碳原子,尤其包含1
‑
20个,优选1
‑
10个,尤其是1
‑
5个和更优选2
‑
3个碳原子的基团,r’和r”具有上述含义,和n是1
‑
10,优选1
‑
4的范围内的整数和更优选是1或2。
9.表述“具有1至30个碳原子的基团”是指具有1至30个碳原子的有机化合物的残基。其不仅包括芳族的和杂芳族的基团,而且包括脂族的和杂脂族的基团,例如烷基、环烷基、烷氧基、环烷氧基、环烷硫基和烯基基团。在此,所述基团可以是支化的或非支化的。
10.根据本发明,芳族基团是优选包含6至20个,更特别6至12个c原子的单或多环芳族
化合物的残基。杂芳族基团是其中至少一个ch基团被n替代和/或至少两个相邻的ch基团被s、nh或o替代的芳基基团。
11.根据本发明优选的芳族或杂芳族基团衍生自苯、萘、联苯、二苯醚、二苯基甲烷、二苯基二甲基甲烷、双苯酮(bisphenon)、二苯砜、噻吩、呋喃、吡咯、噻唑、唑、咪唑、异噻唑、异唑、吡唑、1,3,4
‑
二唑、2,5
‑
二苯基
‑
1,3,4
‑
二唑、1,3,4
‑
噻二唑、1,3,4
‑
三唑、2,5
‑
二苯基
‑
1,3,4
‑
三唑、1,2,5
‑
三苯基
‑
1,3,4
‑
三唑、1,2,4
‑
二唑、1,2,4
‑
噻二唑、1,2,4
‑
三唑、1,2,3
‑
三唑、1,2,3,4
‑
四唑、苯并[b]噻吩、苯并[b]呋喃、吲哚、苯并[c]噻吩、苯并[c]呋喃、异吲哚、苯并唑、苯并噻唑、苯并咪唑、苯并异唑、苯并异噻唑、苯并吡唑、苯并噻二唑、苯并三唑、二苯并呋喃、二苯并噻吩、咔唑、吡啶、联吡啶、吡嗪、吡唑、嘧啶、哒嗪、1,3,5
‑
三嗪、1,2,4
‑
三嗪、1,2,4,5
‑
三嗪、四嗪、喹啉、异喹啉、喹喔啉、喹唑啉、噌啉、1,8
‑
萘啶、1,5
‑
萘啶、1,6
‑
萘啶、1,7
‑
萘啶、酞嗪、吡啶并嘧啶、嘌呤、喋啶或喹嗪、4h
‑
喹嗪、二苯醚、蒽、苯并吡咯、苯并噻二唑(benzooxathiadiazol)、苯并二唑、苯并吡啶、苯并吡嗪、苯并吡嗪烷(pyrazidin)、苯并嘧啶、苯并三嗪、中氮茚、吡啶并吡啶、咪唑并嘧啶、吡嗪并嘧啶、咔唑、吖啶、吩嗪、苯并喹啉、吩嗪、吩噻嗪、吖啶嗪(acridizin)、苯并喋啶、菲咯啉和菲,它们也可任选被取代。
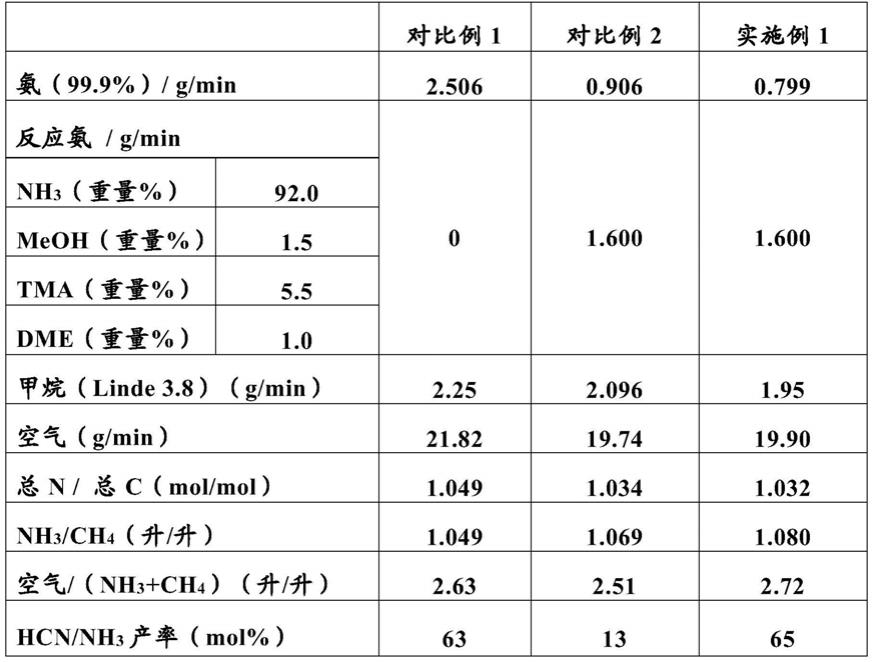
[0012]
优选的烷基基团包括甲基、乙基、丙基、异丙基、1
‑
丁基、2
‑
丁基、2
‑
甲基丙基、叔丁基、戊基、2
‑
甲基丁基、1,1
‑
二甲基丙基、己基、庚基、辛基、1,1,3,3
‑
四甲基丁基、壬基、1
‑
癸基、2
‑
癸基、十一烷基、十二烷基、十五烷基和二十烷基。
[0013]
优选的环烷基基团包括环丙基、环丁基、环戊基、环己基、环庚基和环辛基,它们可任选被支化的或非支化的烷基基团取代。
[0014]
优选的烯基基团包括乙烯基、烯丙基、2
‑
甲基
‑2‑
丙烯基、2
‑
丁烯基、2
‑
戊烯基、2
‑
癸烯基和2
‑
二十碳烯基基团。
[0015]
优选的杂脂族基团包括上述优选的烷基和环烷基基团,其中至少一个碳单元被o、s或基团nr8或nr8r9替代,和r8和r9各自独立地是具有1至6个碳原子的烷基基团、具有1至6个碳原子的烷氧基基团或芳基基团。
[0016]
根据本发明最优选地,所述羧酰胺带有包含1至20个碳原子,优选1至12个,有利地1至6个和特别是1至4个碳原子的支化的或非支化的烷基或烷氧基基团,和包含3至20个碳原子,优选5至6个碳原子的环烷基或环烷氧基基团。
[0017]
所述基团r可带有取代基。优选的取代基尤其包括卤素,特别是氟、氯、溴,以及烷氧基或羟基基团。
[0018]
在本发明的方法中,可单独地或者作为两种或三种或更多种不同的α
‑
羟基羧酰胺的混合物使用所述α
‑
羟基羧酰胺。特别优选的α
‑
羟基羧酰胺包括α
‑
羟基异丁酰胺(hiba)和/或α
‑
羟基丙酰胺。
[0019]
在根据本发明方法的一个变化形式中进一步特别令人感兴趣的是使用可通过氰醇合成得自酮或醛和氢氰酸的那些α
‑
羟基羧酰胺。所述合成中的第一步中使羰基化合物,例如酮,特别是丙酮,或醛,例如乙醛、丙醛、丁醛,与氢氰酸进行反应以提供相应的氰醇。特别优选,所述合成中在作为催化剂的少量的碱或胺的存在下使丙酮和/或乙醛以典型的方式进行反应。在进一步的步骤中,将因此获得的氰醇与水反应以提供所述α
‑
羟基羧酰胺。
[0020]
可成功用于本发明的方法中的醇包括本领域技术人员熟知的任何醇,以及能够在所给出的压力和温度条件下与hca经历醇解型反应的醇的前体化合物。优选通过用优选包含1
‑
10个碳原子,更优选包含1至5个碳原子的醇的醇解进行所述hca的反应。优选的醇尤其是甲醇、乙醇、丙醇、丁醇,特别是正丁醇和2
‑
甲基
‑1‑
丙醇、戊醇、己醇、庚醇、2
‑
乙基己醇、辛醇、壬醇和癸醇以及它们的混合物。特别优选所用的醇是甲醇和/或乙醇,其中甲醇是最有利的。原则上也可以使用醇的前体。因此可以使用例如甲酸烷基酯。甲酸甲酯,或甲醇与一氧化碳的混合物是特别合适的。
[0021]
在本发明的情况下,已经发现描述的操作过程可容忍宽范围的反应物用量比例。因此,可以在相对于所述hca相对高的醇过量或不足量的情况下进行所述醇解。特别优选如下方法变化方案,在该方法变化方案中,所述反应物的转化是在范围为1:3至20:1的醇对hca的摩尔起始比例下进行的。非常特别合适地,所述比例是1:2至15:1,并且甚至更合适地是1:1至10:1。
[0022]
在根据本发明的方法的一个实施方案中,在α
‑
羟基羧酰胺和醇之间的反应是在压力反应器中进行的。为此目的,ep 2018362和wo 2013/026603为了公开的目的通过引用并入本申请中。
[0023]
在1
‑
100巴的压力下进行所述醇解。另外,在从产物混合物中分离/移除氨的过程中,所述压力也大于1巴。更特别地,这意味着在所述反应中形成的氨也在大于1巴的压力下从所述混合物中蒸馏出,其中完全省却了辅助物质的使用,所述辅助物质例如是用于蒸馏移除所述氨的汽提气。
[0024]
为了本发明的目的,不仅贫化了所述产物混合物的氨,而且还贫化了未转化的醇。正是对于将甲醇用于醇解的情况,结果是产物混合物尤其具有氨和甲醇组分,它们原则上非常难以彼此分开。在最简单的情况下,为了使所述产物混合物贫化氨和醇,将所述两种组分从所述产物混合物中作为物质混合物直接移除。然后所述两种物质经历下游的分离操作,例如精馏。另一方面,为了本发明的目的,还可以在一次操作中将所述醇(甲醇)和氨这两种组分从所述产物混合物中分离,和同时,还可以再将所述氨和醇(甲醇)这两种成分彼此分开。
[0025]
在本发明的优选的方法改进中,可能特别令人感兴趣的是,将所述反应步骤和从所述产物混合物中移除所述氨/醇的过程在空间上彼此分开,和在不同的单元中进行。为此目的,可以例如提供一个或多个压力反应器和将它们与压力蒸馏塔组合。这些是布置在所述塔外在分开区域中的一个或多个反应器。
[0026]
在最广义的意义上,这意味着将包含α
‑
羟基羧酰胺和醇作为反应物的反应物料流进料到压力反应器中,使所述反应物料流在1
‑
100巴范围内的压力下在所述压力反应器中彼此进行催化反应,将所得的产物混合物从所述压力反应器排出,和使所述产物混合物贫化醇和氨,其中采用在恒定保持大于1巴的压力下蒸馏移除氨。
[0027]
在该方法变化方案的特定实施方案中,所述反应物的转化和氨/醇的移除在两个不同的在空间上彼此分开的单元中进行。这具有的优点是可采用不同的压力范围用于所述反应物的反应/转化以及氨/醇的随后移除。通过将所述方法分成在比压力塔中的分离步骤更高的压力下的压力反应器中的转化步骤,其中这两个步骤都在超压(即大于1巴)下进行,成功实现再次显著改进所述分离作用和增大移除所述氨/醇混合物的效率。
[0028]
所述品质特征可通过如下方式而再进一步改进:用在所述分离塔(压力蒸馏塔)的底部的方向贫化了氨和醇的产物混合物重复在所述压力反应器中的反应一次或多次,其中反应步骤转移到多个串联的压力反应器。
[0029]
对于所给出的方法变化方案,已经发现在塔和反应器中各种温度范围是合适的。因此,所述压力蒸馏塔通常和优选具有约50℃至约180℃范围内的温度。确切的温度通常通过作为存在的压力条件的函数的沸腾体系调节。在hiba与甲醇的反应中,在所述反应器中的温度优选在约120
‑
240℃的范围内。
[0030]
除了所述的变化方案外,在该变化方案中所述α
‑
羟基羧酰胺与来自在此尤其所得的氨的移除过程中的醇的反应在两个空间上彼此分开但连接的单元中进行,在进一步的方法改进中可优选在单一的单元中进行所述反应步骤和所述移除步骤。压力反应器和压力蒸馏塔在此在单一的单元中实现,似乎同时发生。
[0031]
在根据本发明的方法的另一个变化方案中,将所获得的hce至少部分地,优选至少60重量%,经由气相从所述反应混合物中移除。
[0032]
相应地,该变化方案优选以如下方式实施:将最大比例的所述产物转化成气相。该目的可尤其通过如下方面实现:选择所述反应器,选择压力和温度,和在操作所述反应器时的气体体积,尤其关于所述总体积或所述液体体积。
[0033]
在此可以如下方式实施所述反应:在分开的步骤中将所述hce与由所述反应混合物释放的含氮化合物分开。在特征在于优选与释放的氨一起从所述反应混合物中移除所述hce的实施方案中显现出优点。
[0034]
通过其中在从所述反应混合物中移除这些组分的过程中hce对氨的摩尔比在2:1至1:2的范围内的方法,尤其显现出另外的优点。特别令人感兴趣的是其中将在所述反应混合物的液相中的hce浓度优选保持小于30重量%的方法。优选地,在所述反应混合物的液相中,hce对α
‑
羟基羧酰胺的摩尔比小于1。
[0035]
关于所述方法的生产率的另外的优点可通过将所述醇作为气体引入到所述反应混合物中实现。用于实施本发明方法的反应器的类型不受限制。然而优选使用其中可引入或排出较大气体量的那些反应器。相应地优选使用多相反应器用于实施这个方法变化方案。在此可以使用其中将气体相对于所述液相逆流引入的多相反应器。这些反应器尤其包括基于喷射(begasten)搅拌釜或级联的反应器。另外,所述醇可以气体形式,与所述液体逆流导引通过托盘塔或具有无规填料的塔。
[0036]
在一个优选的实施方案中,可将所述醇顺流引入到所述反应混合物中。为此目的特别合适的反应器尤其包括滴流床反应器、鼓泡塔反应器、喷射洗涤器和降膜反应器,其中特别优选滴流床反应器和降膜反应器,或滴流床反应器和降膜反应器的组合。
[0037]
根据本发明的转化在催化剂的存在下发生。这些包括均相催化剂和非均相催化剂。
[0038]
用于实施根据本发明的方法的示例性均相催化剂是耐水性镧系化合物。镧系化合物是指得自镧系元素,例如la、ce、pr、nd、pm、sm、eu、gd、td、dy、ho、er、tm、yb和/或lu的化合物。优选使用包含镧的镧系化合物。
[0039]
优选地,所述镧系化合物具有在25℃下在水中的溶解度为至少1g/l,优选至少10g/l。
[0040]
优选的镧系化合物是优选处于氧化态为3的盐。
[0041]
除了均相催化的优选变化方案,采用非均相催化剂的方法也是合适的。可成功使用的非均相催化剂尤其包括氧化镁、氧化钙和碱性离子交换剂,和另外的类似实例。
[0042]
例如,优选的方法可以是其中所述催化剂是含有至少一种选自sb、sc、v、la、ce、ti、zr、hf、v、nb、ta、cr、mo、w、tc、re、fe、co、ni、cu、al、si、sn、pb和bi的元素的不溶性金属氧化物的那些。
[0043]
对此另选地,优选的方法可以是其中所用的催化剂是选自ti、zr、hf、v、nb、ta、cr、mo、w、fe、co、ni、cu、ga、in、bi和te的不溶性金属的那些。
[0044]
优选的非均相催化剂尤其包括基于zro2和/或al2o3的催化剂。这种通用类型的特别优选的催化剂尤其详细描述在jp 6
‑
345692中,其中在公开文本jp 06
‑
345692中详述的催化剂为了公开的目的通过引用并入本申请中。
[0045]
其它合适的催化剂描述于2013年7月12日在德国专利商标局提交的de 102013213699中。该公开文本为了公开的目的通过引用并入本申请中。
[0046]
这里特别优选基于zro2和al2o3的催化剂,其中非常特别优选使用氧化镧
‑
、氧化硅
‑
或氧化钇
‑
掺杂的zro2催化剂。后者例如可作为氧化锆催化剂sz 61157商购自saint
‑
gobain nopro公司。在氧化锆晶格中嵌入的钇导致即使在室温下氧化锆的四方相的稳定化,其否则仅在高于1200℃是稳定的。在工业中,它们作为氧导体用于固态氧化物燃料电池或者在氧测量仪(λ探针)中使用。此处具有8mol%的y2o3的组成是典型的。在根据本发明的方法中,使用0.05
‑
20mol%,优选0.5
‑
15mol%,更优选1
‑
10mol%和最优选2
‑
5mol%的基于zro2的氧化镧、氧化硅或氧化钇含量。还可以使用提及的催化剂的混合物。
[0047]
当使用al2o3时,已经发现用bao掺杂是有用的。采用基于al2o3计0.01
‑
1.2mol%的bao获得了良好的结果。特别优选0.05
‑
1.0mol%,非常特别优选0.1
‑
0.8mol%。
[0048]
已经发现,令人惊奇地,这些催化剂对水的存在具有高的耐受性。因此,在所述醇解反应中,在所述反应物进料中的水含量可以是0.1
‑
20mol%。优选0.5
‑
10mol%,特别优选1
‑
3mol%。
[0049]
所述反应温度可在宽范围内变化,其中反应速率通常随着温度提高而增加。温度上限通常由所用醇的沸点确定。优选地,所述反应温度在40
‑
300℃,更优选120
‑
240℃的范围内。
[0050]
在根据本发明的方法的另一个变化方案中,所述醇解可在气相中进行。此处公开的但不限制本发明方法的示例性的气相变化方案是ep 2415750。
[0051]
那里描述了在二氧化锆催化剂的存在下,在150
‑
270℃的温度和1
‑
300kpa的压力下的气相方法,其中所述二氧化锆催化剂还可以含有例如b、al、mn、co、ni、y、la或yb或它们的混合物的元素。
[0052]
该气相方法在固定床或流化床反应器中采用提及的非均相催化剂进行。此处原则上在气相中进行所述反应,其中液相的比例为基于原料总量计的10重量%或更小。
[0053]
可在此在计量到所述反应器中之前蒸发或在所述反应器自身中蒸发醇和hca。另外,可将醇和hca分开地或者以已经混合的形式进料到所述反应器。优选其中在惰性气体,优选氮下进行所述反应的变化方案,这使得可以更容易地蒸发,因为所述反应组分的分压降低。
[0054]
当在所述反应器中蒸发所述反应组分时,可以将它们与溶剂一起计量到所述反应器中。可能的溶剂例如是基于醚的溶剂,例如四氢呋喃,基于酰胺的溶剂,例如n
‑
甲基吡咯烷酮,或基于酯的溶剂,例如乳酸甲酯,或类似物。然而对于该变化方案,优选无溶剂实施所述气相反应。
[0055]
相应地选择反应温度,使得所述反应组分以充分蒸发的形式存在于所述反应器中。这取决于所述酰胺或醇的性质,其摩尔比例,和惰性气体或溶剂的存在。为了充分蒸发hiba,选择>150℃的反应温度,和在大气压力下进行反应的情况下,反应温度为>180℃。如果将所述反应温度保持为<240℃,则避免了hiba分解成丙酮,或者形成副产物例如α
‑
烷氧基异丁酸或通过脱氢形成的烯烃衍生物。
[0056]
为了获得在长时间内稳定的转化率,计量速率为基于所用的催化剂量计0.01
‑
5重量份/小时的hca。优选地,基于所述醇反应组分的whsv(重时空速)为0.01
‑
100h
‑1。
[0057]
为了排出想要的hce,和为了将其与氨、形成的副产物或未转化的起始产品分开,在该方法变化方案中可以使用标准分离方法,例如蒸馏。
[0058]
根据本发明的另一个气相方法变化方案在水的存在下进行。已经发现,令人惊奇地,例如在hiba与甲醇在水的存在下进行反应的情况下,非常显著地抑制了副产物的形成,所述副产物尤其是丙酮或2
‑
氨基
‑2‑
甲基丙腈(ampn),并且mhib的选择性和催化剂寿命被显著提高。水可以被添加到所述反应物进料中或者直接进料到所述反应器中。水对hca的摩尔比为0.1
‑
10,优选0.3
‑
5和更优选0.5
‑
1mol/mol。
[0059]
在该变化方案中醇对hca的摩尔比为1
‑
25,优选3
‑
20和更优选5
‑
9mol/mol。
[0060]
所述hca的醇解中产生大量的副产物,尤其是烷基胺和烯烃,它们难以与所述反应产物氨分离。如果在所述优选的变化方案中,所述醇解采用所述反应组分hiba和甲醇进行,这种甲醇解中形成作为副产物的二甲氧基丙烷、甲氧基丙烯、甲酸甲酯、乙酸甲酯、二甲基异丙基胺、丙烯和尤其是二甲胺和三甲胺。
[0061]
如果这种反应氨,在与未转化的醇分开之后,作为起始产品被直接进料到hcn工艺中,例如进料到安德卢梭(andrussow)工艺中,则在极短的时间后,在数分钟内,就已经发生催化剂活性的显著下降,这可以从在催化剂网处的温度的明显提高注意到。
[0062]
现在已经发现,令人惊奇地,在中间连接有包括固体吸附剂的纯化步骤的情况下,可避免这种在催化剂活性方面的下降,尽管至少烷基胺仍存在于进料到所述hcn工艺中的氨中。在这种情况下,所述烷基胺杂质的基于氨的浓度可以是0.1
‑
10重量%,优选0.2
‑
8重量%和更优选0.5
‑
6重量%。
[0063]
对于根据本发明的纯化步骤有用的固体吸附剂优选是活性炭。
[0064]
活性炭可以所有可能的形态形式使用,以粉末形式,以颗粒化形式,或者作为圆柱体或球形粒料。优选具有1000
‑
1500m2/g,更优选1200
‑
1400m2/g的表面积值的颗粒化和粒料化的活性炭。除了用氯化锌或磷酸进行化学活化的活性炭外,还优选已经使用碱金属盐、碱金属、氯化物、硫酸盐和乙酸盐进行气体活化的活性炭。
[0065]
特别合适的活性炭是如下可商购自donau carbon公司的hydraffin cc 12
×
40,alcarbon dc 60/8
×
16或supersorbon c iv。
[0066]
可能的根据本发明的吸附器是固定床、移动床或流化床吸附器,在此优选前者。示例性的装置方案例如描述在ullmann's encyclopedia of industrial chemistry(乌尔曼
工业化学大全),wiley 2012,第555页及后续页(doi:10.1002/14356007.b03_09.pub2)中。所述操作过程可以连续或间歇进行,优选前者。
[0067]
在0
‑
150℃,优选30
‑
100℃,更优选60
‑
80℃的温度范围内,和在0.05
‑
5巴,优选0.2
‑
4巴,更优选在1
‑
3.5巴的压力下进行所述吸附。
[0068]
根据本发明纯化的氨可因此在各种hcn工艺或其它制备工艺中毫无问题地用作反应物。例如,在ep 0941984中详述了氨与甲醇反应得到hcn的反应。另外,通过bma或安德卢梭工艺,可由氨和甲烷得到hcn,在此这些工艺描述于ullmann’s encyclopedia of industrial chemistry,第5版,cd
‑
rom版,关键词“无机氰基化合物(inorganic cyano compounds)”。同样可能将氨例如再循环到氨氧化工艺中,例如从氨、氧和丙烯进行的丙烯腈的工业级合成。丙烯腈合成例如描述于k.weisermehl和h.
‑
j.arpe的industrial organic chemistry(工业有机化学),在关键词“sohio工艺”部分中,在第307页及后续页。
[0069]
具体地,本发明提供以下技术方案:
[0070]
项目1.从氢氰酸出发制备α
‑
羟基羧酸酯的方法,其特征在于将在相应的α
‑
羟基羧酰胺的醇解步骤中形成的氨,在纯化步骤后,在仍包含至少一种烷基胺的情况下再循环到通过bma或安德卢梭工艺进行的氢氰酸制备工艺中,或再循环到氨氧化工艺中用于制备丙烯腈。
[0071]
项目2.根据项目1的方法,其特征在于基于氨的烷基胺总量的浓度为1
‑
100 000ppm。
[0072]
项目3.根据项目1的方法,其特征在于所述α
‑
羟基羧酸酯是羟基异丁酸甲酯。
[0073]
项目4.根据项目1的方法,其特征在于所述烷基胺是三甲胺。
[0074]
项目5.根据项目1的方法,其特征在于所述氢氰酸制备根据安德卢梭法进行。
[0075]
项目6.根据项目1的方法,其特征在于通过导引经过固体,以吸附方式纯化所述氨。
[0076]
项目7.根据项目1的方法,其特征在于在连续操作的吸附床中纯化所述氨。
[0077]
项目8.根据项目1的方法,其特征在于采用活性炭纯化所述氨。
[0078]
项目9.根据项目1的方法,其特征在于所述羟基羧酰胺的醇解反应在液相中或在气相中进行。
[0079]
项目10.根据项目1的方法,其特征在于所述纯化在0℃至150℃的温度范围内进行。
[0080]
项目11.根据项目1的方法,其特征在于所述纯化在0.05至5巴的压力范围内进行。
[0081]
项目12.根据项目1的方法,其特征在于
[0082]
a)将包含α
‑
羟基羧酰胺和醇的反应物料流进料到含有催化剂的反应器中,
[0083]
b)将该反应混合物在压力反应器中在0.1
‑
100巴范围内的压力下彼此进行反应,
[0084]
c)使得自b)的产物混合物贫化醇和氨,
[0085]
d)将醇和至少含有三甲胺的氨分开,和
[0086]
e)在计量加到氢氰酸制备工艺中之前,通过活性炭纯化至少含有三甲胺的氨。
具体实施方式
[0087]
下面的实施例意于示例性说明根据本发明的方法,但不以丝毫方式限制它。
[0088]
在如图1中示出的装置中进行所述实施例和对比例。通过三个计量线路1、2和3,将氨、空气和甲烷引入到所述试验装置中。可以通过两个三通阀(dw)直接或经由用活性炭填充的吸附床(a)将氨进料到静态搅拌器(b)中。所述吸附床是可加热的。将所述反应物在静态混合器中混合和然后经由线路8进料到预热器(c),其被预热到希望的温度并通过线路9引入到反应器(r)中。后者配备有催化剂网(d)和空气冷却器(e);将产物气体混合物在后者中冷却到希望的温度。然后将该产物气体混合物部分经由线路12送去在线分析,或部分经由线路11送去燃烧,因为在所述试验的情况下没有预先设计对所形成的hcn的贮存。
[0089]
附图标记列表:
[0090]
1:氨引入线路
[0091]
2:空气引入线路
[0092]
3:甲烷引入线路
[0093]
4:吸附床的旁路线路
[0094]
5:吸附器引入线路
[0095]
6:吸附器引出线路
[0096]
7:静态混合器引入线路
[0097]
8:预热器引入线路
[0098]
9:安德卢梭反应器引入线路
[0099]
10:安德卢梭反应器引出线路
[0100]
11:产物混合物的部分引出线路
[0101]
12:在线分析引入线路
[0102]
13:在线分析引出线路
[0103]
14:产物混合物总引出线路
[0104]
a:吸附床
[0105]
b:静态混合器
[0106]
c:预热器
[0107]
d:催化剂网
[0108]
e:空气冷却器
[0109]
f:在线分析
[0110]
r:反应器
[0111]
对比例1
‑
2,实施例1:
[0112]
这些在类似于图1的装置中进行。在对比例1中,使用纯氨;在对比例2中,向其中添加34重量%的得自hiba醇解的反应氨和催化剂。在这两种情况下,将所述氨进料不引导通过所述吸附床。在本发明实施例1中,重复对比例2,但是使全部氨进料导引通过所述吸附床,所述吸附床被填充以hydrafin cc 12
×
40活性炭。对于对比例1和实施例1在14天的tos(运行时间)后,和对于对比例2在1小时tos后,结果示于表1中。同样示出的是在引入到所述吸附床中之前,在所述反应氨中的胺杂质。
[0113]
表1:活性炭的影响
[0114][0115]
采用活性炭纯化的含有反应氨的氨进料,与未纯化的相比,显著增加了hcn的产率,并且与纯氨相当。
[0116]
实施例2
‑
4:
[0117]
重复实施例1,并在各个不同吸附床温度下进行所述吸附。在此所用的活性炭是得自donau carbon公司的alcarbon ph 55
×
8c。结果示于表2中。
[0118]
表2:温度依赖性
[0119] 实施例2实施例3实施例4吸附床温度(℃)20℃70℃100℃nh3(重量%)95.78395.08495.084tma(重量%)2.0972.5122.512dme(重量%)0.7920.7530.753meoh(重量%)1.3281.651.65hcn/nh3产率mol%63.2662.5762.51
[0120]
用活性炭进行的处理实现了在宽温度范围内在hcn产率方面的良好结果。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1