一种烯效唑(E)-(S)-对映体的不对称批量合成方法与流程
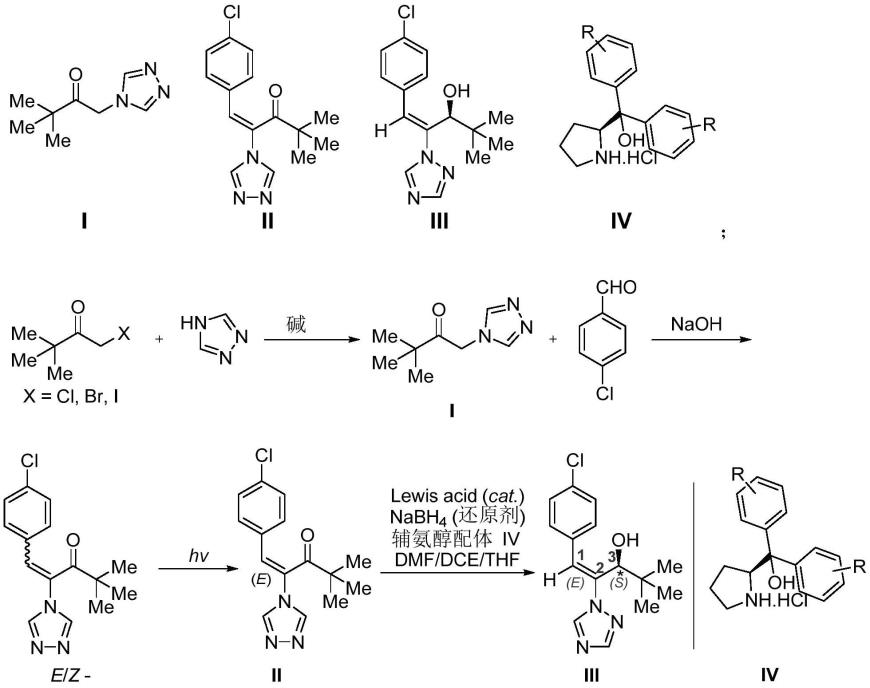
一种烯效唑(e)-(s)-对映体的不对称批量合成方法
技术领域
1.本发明属于化学制备技术领域,具体涉及一种烯效唑(e)-(s)-对映体的不对称批量合成方法。
背景技术:
2.植物体的生长与手性化合物存在着非常密切的联系。一方面,植物分泌、合成的一些手性化合物,如糖苷、酶、萜类、有机酸及植物激素等化合物,在植物的生理生化过程中起着重要的作用;另一方面,人工合成的手性化合物,尤其是农药等与植物具有对映体选择性相互作用,它们或是选择性地抑制植物体的生长,或是被其选择性地吸收和利用。大多数三唑类植物生长调节剂具有两个或者两个以上的对映异构体。之前的研究工作显示,三唑类植物生长调节剂的生物活性具有对映选择性。例如常见的植物生长调节剂烯效唑,其s体对于抑制植物生长的活性是r体的7倍,张玉芬的研究显示抑芽唑s对映体抑制大麦生长的活性比r对映体要强得多,lenton等人也发现多效唑2s,3s-对映体对小麦苗茎的抑制作用要比2r,3r-对映体强。
3.烯效唑是上个世纪80年代研制成功的一类广谱且高效的三唑类植物生长调节剂,它被植物吸收后,可以阻碍内源赤霉素的生物合成,从而延缓植株的营养生长,减缓细胞的分裂和伸长,抑制茎秆伸长,幼苗高度明显降低,茎粗和分蘖数增加,最终达到作物增效增产的目的。从烯效唑的构造式可以看出,l,2-碳碳双键分别连接着不同的原子和原子团,说明烯效唑具有z-型、e-型异构体,而3号与羟基相连的碳原子为手性碳,说明烯效唑具有r-、s-型旋光异构体。烯效唑的能量有利的构象异构体主要有e-s、e-r、z-s、z-r、e-rs、z-rs等,这些异构体杀菌和控长的生理效应有着明显的差别[(1)陈学恒,刘晓庚,夏红英等.江西化工,1998,3:9-16.(2)刘秀娣,黄华强.江西农药,1993,4:10-13.]。研究表明烯效唑的不同异构体的活性次序为:植物生长调节活性:e-s》e-rs》z-rs≥e-r;抑菌活性:e-r》e-rs》z-rs≥e-s。e-异构体表现出优异的生物活性,而z-异构体在很大程度上无活性。
[0004]
烯效唑(penxefezol),学名为(e)-(rs)-1-(4-氯苯基)-4,4-二甲基-2-(1h-1,2,4-三唑-1-基)戊-1-烯-3-醇,是继植物生长调节剂多效唑(pp333)之后由日本住友化学公司开发的又一高活性的新型植物生长调节剂,并兼有卓越的杀菌效果,也是目前所开发的唑类农用化学品中矮化植株、抗倒伏能力最强的植物生长调节剂[(1)fletcher r a,hofstra g,gao jian-guo.plant cell physiol,1986,27(2):367-371.(2)李煜昶,成俊然,郑健禺.农药,1993,32(6):11-13.(3)paul a.worthingto.pestic sci,1991,31:457-498.]。研究结果表明,烯效唑的(e)-(r)-(-)-对映体具有良好的杀菌活性,且(e)-(s)-(+)-对映体具有良好的植物生长调节活性和除草活性。由于手性农药对映体功效和毒性不同,对其对映体进行拆分、测定含量很有必要。在此基础上,应用活性较高的单对映体,可使化学物质用量减少,从而使实际应用过程的经济性提高,对环境污染的减少和安全性的提高具有重要的意义。
[0005]
目前已经报道了两种烯效唑的(e)-(s)-(-)-对映体的合成方法。一种是通过生成
对映体酯来拆分:外消旋体与具有光学活性的羧酸在碱存在下反应得到一对非对映体酯,烯效唑的(e,s)-(-)-或(e,r)-(+)-对映体是通过拆分和分解非对映体酯混合物获得的[priesnitz,u.german patent 3515869,1986;chem.abstr.1987,106,50229.]。另一种方法是通过相应的酮的不对称还原。具体的是在手性配体存在下的不对称还原为相应的酮,用氢化铝锂[yoneyoshi,y.;sazukamo,g.;hamada,k.;nishioka,t.european patent 171175,1986]或硼氢化钠(钾)还原酮[yoneyoshi,y.;suzukamo,g.;sakito,y.;nishioka,t.pct int.appl.wo 8504401,1985]。
[0006]
在不加手性辅助剂作为还原试剂的前提下,利用氢化铝锂或硼氢化钠(钾)还原烯唑酮前体只能得到1:1的对映体,ee值为0。为了改善还原反应特性,增大(e)-(s)-对映体的产量,必须添加适量的具有特殊结构的手性辅助试剂已改善其ee值。由于经济和安全因素,文献报道的方法中常用的还原剂是硼氢化钠(钾),其中手性配体的设计和合成是硼氢化还原反应中最重要的因素。yoneyoshi利用硼氢化钠作为还原试剂,通过手性氨基醇盐酸盐作为辅助试剂,应用于α,β-不饱和三唑酮前体的不对称还原,可以得到良好转化率和低至中等对映选择性的光学活性物质(yoneyoshi,y.;suzukamo,g.;sakito,y.;nishioka,t.pct int.appl.wo 8403885,1984)。liao报道了在利用手性相转移催化剂(chiral pta)存在下,通过硼氢化钾还原α,β-不饱和三唑酮前体的反应,最高可以获得92%的收率和30%ee的光学活性烯效唑的(e)-(s)-(-)-对映体(liao,l.a.;cui,h.x.;chen,m.d.;zhang,h.k.;guo,q.z.;chin.j.app.chem.1998,15,39)。zhou等人报道了利用硼氢化钠作为还原试剂,通过手性脯氨醇盐酸盐作为辅助试剂,应用于α,β-不饱和三唑酮前体的不对称还原,可以得到良好转化率和中等至优秀对映选择性的光学活性物质(zhou z.;tang y.;wang l.;et al.synthesis,2004,2:217
–
220.)。
[0007]
以上这些方法,为合成烯效唑(e)-(s)-对映体提供了可供选择的途径。实际生产中发现,上述方案对于实验室小规模克级以下制备时可行,然而对于工业级大规模制备的时候分别存在着如下几个问题:(1)还原剂使用量大,导致烯唑酮原料中的烯基双键容易被过渡还原;(2)实际得到的ee值降低厉害,且工艺不稳定,(3)所用溶剂量较大,1mol底物需要12l dce和6l dmf;(4)辅助试剂l-脯氨醇用量较大,相对于底物烯酮需1.5eq。基于手性烯效唑所具有的强效的农药活性,我们预计开发有效的方法合成含有该结构特征的化合物将具有十分强大的可转化能力,以及十分广阔的工业级应用前景。
技术实现要素:
[0008]
发明目的:本发明目的在于针对现有技术的不足,提供一种烯效唑(e)-(s)-对映体的不对称批量合成方法,该方法对映选择性高,产率高,操作简便,副产物少,适用于较大规模的制备。
[0009]
技术方案:本发明所述的一种一种烯效唑(e)-(s)-对映体的不对称批量合成方法,包括如下步骤:
[0010]
(1)冰浴条件下,在有机溶剂中,将3,3-二甲基-1-卤代-2-丁酮与1,2,4-三氮唑作混合,然后撤出冰浴,体系逐渐加温至室温搅拌反应2-2.5h,合成结构如式i化合物的3,3-二甲基-1-(1,2,4-三氮唑)-2-丁酮中间体;
[0011]
(2)将式i化合物3,3-二甲基-1-(1,2,4-三氮唑)-2-丁酮与对氯苯甲醛在无机碱
的作用下发生缩合反应,并同时在紫外光照射条件下进行转位反应,合成结构如式ii化合物(e)-烯唑酮中间体;
[0012]
(3)将式ii化合物(e)-烯唑酮在路易斯酸催化剂和辅氨醇手性辅助试剂iv的共同作用下,与金属硼氢化物发生还原反应,反应温度为45-50℃,反应时间为6~8小时,反应溶剂为有机混合溶剂,对映选择性地合成得到结构如式iii的产物烯效唑(e)-(s)-对映体;
[0013]
(4)反应完毕,洗涤反应液,分离得到目标产物烯效唑(e)-(s)-对映体,并回收辅氨醇手性辅助试剂iv;
[0014][0015]
其中:式iv中r为h或供电子基团烷基、烷氧基、环烷基或吸电子基团氟、氯、溴、碘、酯基、硝基、氰基、酰胺基或呋喃基、噻吩基、吡啶基、烯基、炔基、硅基。
[0016]
进一步地,作为较优实施方式,所述3,3-二甲基-1-卤代-2-丁酮中的卤素为氯、溴或碘。
[0017]
进一步地,作为较优实施方式,步骤(2)中所述无机碱为碳酸钾、碳酸铯、磷酸钾、三乙胺或dbu中的一种或多种混合物。
[0018]
进一步地,作为较优实施方式,步骤(2)中所述无机碱为氢氧化钠与三乙胺按摩尔比1:1的混合物。
[0019]
进一步地,作为较优实施方式,步骤(1)中所述有机溶剂为甲醇、乙醇、丙醇、正丁醇或叔丁醇中的至少一种。
[0020]
进一步地,作为较优实施方式,所述有机溶剂为甲醇与水或乙醇与水的混合液。
[0021]
进一步地,作为较优实施方式,步骤(3)中所述金属硼氢化物为为硼氢化锂、硼氢化钠、硼氢化钾、硼氢化镁、硼氢化锌或硼氢化铜中的至少一种;所述路易斯酸催化剂为氯
化镁、氯化铜、氯化锌、三氯化铁、氯化亚铁、氯化亚铜、溴化镁、溴化铜、溴化锌或三溴化铁中的至少一种。
[0022]
进一步地,作为较优实施方式,步骤(3)中所述有机混合溶剂为n,n-二甲基甲酰胺、四氢呋喃以及二氯甲烷的混合溶剂或n,n-二甲基甲酰胺、四氢呋喃、二氯甲烷叔戊醇以及三氯甲烷的混合溶剂或n,n-二甲基乙酰胺、四氢呋喃以及二氯甲烷的混合溶剂或n,n-二甲基乙酰胺、1,4-二氧六环以及二氯甲烷的混合溶剂或n,n-二甲基乙酰胺、1,4-二氧六环以及三氯甲烷的混合溶剂。
[0023]
进一步地,作为较优实施方式,步骤(4)中分离目标产物的具体过程为:反应完毕后用60-65℃的热水洗涤反应液,保持60-65℃静置分层,分离水相,有机相再次用60-65℃的饱和食盐水洗涤,分层;分别合并有机相和水相;有机相减压浓缩回收有机溶剂,剩余物重结晶既得目标产品;水相用naoh中和至ph9-10,再次用氯仿萃取,分层,分离回收得到辅氨醇手性辅助试剂iv。
[0024]
有益效果:(1)起始反应物3,3-二甲基-1-卤代-2-丁酮与1,2,4-三氮唑的摩尔比例为1:1就能非常顺利地进行,所用溶剂为污染小的醇类溶剂,以几乎定量的产率得到中间体i;中间体i与对氯苯甲醛反应的原料摩尔数比例为1:1,所用溶剂为醇类与水的混合溶剂,产率达90%以上;此两步反应的产率高,副产物少,所用溶剂为绿色溶剂,这不仅便于产物的分离提纯,而且体现出良好的绿色化。
[0025]
(2)此外,在羰基还原的反应中,通过添加路易斯酸催化剂活化羰基,弱化了硼氢化物的反应性,从而成功抑制了烯基被还原的副反应,提高了主反应还原的化学选择性,通过加入手性辅氨醇衍生物iv作为手性诱导试剂,提高了羰基还原反应的对映选择性,最终可使还原的产率达到95%,ee值达90%以上。
[0026]
(3)反应完毕,可以通过热水洗涤萃取的方式回收手性辅氨醇衍生物iv。通过循环利用手性辅氨醇衍生物iv,可有效降低生产成本。
[0027]
(4)本发明所提供的发明方法底物便宜易得,各步反应产率高,副产物少,条件温和,溶剂多选低毒污染小的醇类溶剂,可以回收重复利用,反应的化学和对映选择性高,操作简单,成本极低。最终目标产品烯效唑(e)-(s)-对映体产率可到95%,ee值达90%以上,表明该反应可适用于较大规模的制备,在农药生产等领域具有非常好的应用前景。
附图说明
[0028]
图1为实施例1中产物烯效唑(e)-(s)-对映体的1hmr谱图;
[0029]
图2为实施例1中烯效唑(e)-(s)-对映体的ee值谱图;
[0030]
图3为实施例8中烯效唑(e)-(s)-对映体的ee值谱图。
具体实施方式
[0031]
下面通过附图对本发明技术方案进行详细说明,但是本发明的保护范围不局限于所述实施例。
[0032]
实施例1:
[0033]
冰水浴下将1.34g的3,3-二甲基-1-氯-2-丁酮与0.69g的1,2,4-三氮唑混合于冰冷的无水乙醇(10ml)中,加入碳酸钾1.38g。混合均匀后,撤除冰水浴,使体系缓慢升温至室
温,搅拌反应2小时。反应完毕,减压回收乙醇溶剂,水洗反应物,甲苯萃取三次,浓缩甲苯,使产品重结晶析出。过滤即得1.65g的产物3,3-二甲基-1-(1,2,4-三氮唑)-2-丁酮,产率98%。
[0034]
冰水浴下,将0.84g化合物3,3-二甲基-1-(1,2,4-三氮唑)-2-丁酮与0.70g对氯苯甲醛溶于12ml乙醇中,剧烈搅拌下将反应液搅拌冷却至0℃。随后,将0.20g naoh溶于2ml水中,并通过滴液漏斗将该naoh的水溶液缓慢滴加到冷却到0度的乙醇溶液中,控制滴加的温度为0-5℃。滴加完毕,撤除冰水浴,将反应液缓慢升温至室温,继续搅拌反应8小时。反应完毕,浓缩回收乙醇溶剂,过滤收集黄色的固体产物。所得的固体产物重新溶解于无水甲醇中,浓缩重结晶,得到(e/z)-烯唑酮产物(e/z)-1-(4-氯苯基)-4,4-二甲基-2-(4h-1,2,4-三氮唑-4-基)-1-戊烯-3-酮为黄色针状晶体,产品重量1.31g,产率91%。将该(e/z)-烯唑酮针状晶体混合产物重新溶解于无水氯仿中,氮气保护,紫外光照射条件下,室温反应12小时,使(e/z)-烯唑酮全部转位转化为(e)-1-(4-氯苯基)-4,4-二甲基-2-(4h-1,2,4-三氮唑-4-基)-1-戊烯-3-酮,产率100%。
[0035]
氮气保护下,投入(s)-吡咯啉-2-基-1,1,-二对甲苯基甲醇盐酸盐(0.143g.)、nabh4(17mg)、0.5ml二氯乙烷,以及氯化镁(5.76mg)作为路易斯酸催化剂,放入-20℃的冷阱中30min,在搅拌下,2h内将试管中的溶液温度逐渐由-20℃升至室温。之后放入50℃油浴中,投入(e)-1-(4-氯苯基)-4,4-二甲基-2-(4h-1,2,4-三氮唑-4-基)-1-戊烯-3-酮(0.13g)和0.5mldce,tlc监测反应进度。50℃反应4-8h。停止反应,用加入2n hcl水溶液(5ml)淬灭反应并继续搅拌2h。50℃分层,水层用dcm 5x 1.0ml萃取,合并的有机相用饱和食盐水洗涤,无水硫酸钠干燥。浓缩溶液,爬小板分出产物点用于测试ee值,如图3所示。剩余样重结晶,得到产物烯效唑(e)-(s)-对映体iii,129mg,产率91%,ee 76%。
[0036]
如图2所示,是产物iii的核磁共振实验数据:
[0037]1h nmr(400mhz,cdcl3)δ0.59(s,1h),4.27(d,j=8hz,1h),4.47(d,j=12hz,1h),6.84(s,1h),7.24(d,j=8hz,2h),7.32(d,j=8hz,2h),7.98(s,1h),8.43(s,1h).
[0038]
ee值测试条件:仪器agilent 1260infinity;v异丙醇:v正己烷=3:97;流速0.750ml/min;波长230nm;od3柱子。在此条件下,出峰时间分别为22.887min和23.875min。
[0039]
实施例2:
[0040]
本实施例采用实施例1中制得的(e)-烯唑酮中间体,氮气保护下,向(e)-烯唑酮中间体中投入(s)-吡咯啉-2-基-1,1,-二对甲苯基甲醇盐酸盐(1.43g)、nabh4(0.170g)、5.0ml甲苯,以及氯化镁(57.6mg)作为路易斯酸催化剂,放入-20℃的冷阱中30min,在搅拌下,2h内将试管中的溶液温度逐渐由-20℃升至室温。之后放入50℃油浴中,投入(e)-1-(4-氯苯基)-4,4-二甲基-2-(4h-1,2,4-三氮唑-4-基)-1-戊烯-3-酮(1.30g)和5.0ml甲苯,tlc监测反应进度。50℃反应4-8h。停止反应,用加入2n hcl水溶液(10ml)淬灭反应并继续搅拌2h。50℃分层,水层用dcm 5x8 ml萃取,合并的有机相用饱和食盐水洗涤,无水硫酸钠干燥。浓缩溶液,爬小板分出产物点用于测试ee值。剩余样重结晶,得到产物iii,1.31g,产率91%,ee 76%。
[0041]
实施例3:
[0042]
冰水浴下将6.70g的3,3-二甲基-1-氯-2-丁酮与1.40g的1,2,4-三氮唑混合于冰冷的无水乙醇(50ml)中,加入碳酸钾6.90g。混合均匀后,撤除冰水浴,使体系缓慢升温至室
温,搅拌反应2小时。反应完毕,减压回收乙醇溶剂,水洗反应物,甲苯萃取三次,浓缩甲苯,使产品重结晶析出。过滤即得产物i,产品重量8.25g,产率98%。
[0043]
冰水浴下,将8.20g化合物i与7.0g对氯苯甲醛溶于120ml乙醇中,剧烈搅拌下将反应液搅拌冷却至0℃。随后,将2.0g naoh溶于20ml水中,并通过滴液漏斗将该naoh的水溶液缓慢滴加到冷却到0度的乙醇溶液中,控制滴加的温度为0-5℃。滴加完毕,撤除冰水浴,将反应液缓慢升温至室温,继续搅拌反应8小时。反应完毕,浓缩回收乙醇溶剂,过滤收集黄色的固体产物。所得的固体产物重新溶解于无水甲醇中,浓缩重结晶,得到的(e/z)-1-(4-氯苯基)-4,4-二甲基-2-(4h-1,2,4-三氮唑-4-基)-1-戊烯-3-酮产物为黄色针状晶体,产品重量12.4g,产率90%。将该(e/z)-烯唑酮针状晶体混合产物重新溶解于无水氯仿中,氮气保护,紫外光照射条件下,室温反应12小时,使(e/z)-烯唑酮全部转位转化为(e)-1-(4-氯苯基)-4,4-二甲基-2-(4h-1,2,4-三氮唑-4-基)-1-戊烯-3-酮,产率100%。
[0044]
氮气保护下,投入(s)-吡咯啉-2-基-1,1,-二对甲苯基甲醇盐酸盐(7.15g)、nabh4(0.85g)、25ml二氯乙烷,以及氯化铜(330mg)作为路易斯酸催化剂,放入-20℃的冷阱中30min,在搅拌下,2h内将试管中的溶液温度逐渐由-20℃升至室温。之后放入50℃油浴中,投入(e)-1-(4-氯苯基)-4,4-二甲基-2-(4h-1,2,4-三氮唑-4-基)-1-戊烯-3-酮(6.50g)和25ml dce,tlc监测反应进度。50℃反应4-8h。停止反应,用加入2n hcl水溶液(25ml)淬灭反应并继续搅拌2h。50℃分层,水层用dcm 5x50ml萃取,合并的有机相用饱和食盐水洗涤,无水硫酸钠干燥。浓缩溶液,爬小板分出产物点用于测试ee值。剩余样重结晶,得到产物iii,6.01g,产率93%,ee 78%。
[0045]
实施例4:
[0046]
冰水浴下将67g的3,3-二甲基-1-氯-2-丁酮与14g的1,2,4-三氮唑混合于冰冷的无水乙醇(50ml)中,加入碳酸钾69g。混合均匀后,撤除冰水浴,使体系缓慢升温至室温,搅拌反应2小时。反应完毕,减压回收乙醇溶剂,水洗反应物,甲苯萃取三次,浓缩甲苯,使产品重结晶析出。过滤即得产物i,产品重量82.5g,产率98%。
[0047]
冰水浴下,将82g化合物i与70g对氯苯甲醛溶于1200ml乙醇中,剧烈搅拌下将反应液搅拌冷却至0℃。随后,将20g naoh溶于200ml水中,并通过滴液漏斗将该naoh的水溶液缓慢滴加到冷却到0度的乙醇溶液中,控制滴加的温度为0-5℃。滴加完毕,撤除冰水浴,将反应液缓慢升温至室温,继续搅拌反应8小时。反应完毕,浓缩回收乙醇溶剂,过滤收集黄色的固体产物。所得的固体产物重新溶解于无水甲醇中,浓缩重结晶,得到的(e/z)-1-(4-氯苯基)-4,4-二甲基-2-(4h-1,2,4-三氮唑-4-基)-1-戊烯-3-酮产物为黄色针状晶体,产品重量124g,产率90%。将该(e/z)-烯唑酮针状晶体混合产物重新溶解于无水氯仿中,氮气保护,紫外光照射条件下,室温反应12小时,使(e/z)-烯唑酮全部转位转化为(e)-1-(4-氯苯基)-4,4-二甲基-2-(4h-1,2,4-三氮唑-4-基)-1-戊烯-3-酮,产率100%。
[0048]
氮气保护下,投入(s)-吡咯啉-2-基-1,1,-二对甲苯基甲醇盐酸盐(71.5g)、nabh4(0.85g)、250ml二氯乙烷,以及氯化铜(3.3g)作为路易斯酸催化剂,放入-20℃的冷阱中30min,在搅拌下,2h内将试管中的溶液温度逐渐由-20℃升至室温。之后放入50℃油浴中,投入(e)-1-(4-氯苯基)-4,4-二甲基-2-(4h-1,2,4-三氮唑-4-基)-1-戊烯-3-酮(65g)和25ml dce,tlc监测反应进度。50℃反应4-8h。停止反应,用加入2n hcl水溶液(250ml)淬灭反应并继续搅拌2h。50℃分层,水层用dcm 5x500 ml萃取,合并的有机相用饱和食盐水洗
涤,无水硫酸钠干燥。浓缩溶液,爬小板分出产物点用于测试ee值。剩余样重结晶,得到产物iii,60.1g,产率93%,ee 78%。
[0049]
实施例5:
[0050]
冰水浴下将670g的3,3-二甲基-1-氯-2-丁酮与140g的1,2,4-三氮唑混合于冰冷的无水乙醇(500ml)中,加入碳酸钾690g。混合均匀后,撤除冰水浴,使体系缓慢升温至室温,搅拌反应2小时。反应完毕,减压回收乙醇溶剂,水洗反应物,甲苯萃取三次,浓缩甲苯,使产品重结晶析出。过滤即得产物i,产品重量825g,产率98%。
[0051]
冰水浴下,将820g化合物i与700g对氯苯甲醛溶于3l乙醇中,剧烈搅拌下将反应液搅拌冷却至0℃。随后,将200g naoh溶于1.0l水中,并通过滴液漏斗将该naoh的水溶液缓慢滴加到冷却到0度的乙醇溶液中,控制滴加的温度为0-5℃。滴加完毕,撤除冰水浴,将反应液缓慢升温至室温,继续搅拌反应8小时。反应完毕,浓缩回收乙醇溶剂,过滤收集黄色的固体产物。所得的固体产物重新溶解于无水甲醇中,浓缩重结晶,得到的(e/z)-1-(4-氯苯基)-4,4-二甲基-2-(4h-1,2,4-三氮唑-4-基)-1-戊烯-3-酮产物为黄色针状晶体,产品重量1240g,产率90%。将该(e/z)-烯唑酮针状晶体混合产物重新溶解于无水氯仿中,氮气保护,紫外光照射条件下,室温反应12小时,使(e/z)-烯唑酮全部转位转化为(e)-1-(4-氯苯基)-4,4-二甲基-2-(4h-1,2,4-三氮唑-4-基)-1-戊烯-3-酮,产率100%。
[0052]
氮气保护下,投入(s)-吡咯啉-2-基-1,1,-二对甲苯基甲醇盐酸盐(715g)、nabh4(85g)、500ml二氯乙烷,以及氯化锌(38g)作为路易斯酸催化剂,放入-20℃的冷阱中30min,在搅拌下,2h内将试管中的溶液温度逐渐由-20℃升至室温。之后放入50℃油浴中,投入(e)-1-(4-氯苯基)-4,4-二甲基-2-(4h-1,2,4-三氮唑-4-基)-1-戊烯-3-酮(650g)和2.5l dce,tlc监测反应进度。50℃反应4-8h。停止反应,用加入2n hcl水溶液(1.0l)淬灭反应并继续搅拌2h。50℃分层,水层用dcm 5x 2000ml萃取,合并的有机相用饱和食盐水洗涤,无水硫酸钠干燥。浓缩溶液,爬小板分出产物点用于测试ee值。剩余样重结晶,得到产物iii,580g,产率90%,ee 75%。
[0053]
实施例6:
[0054]
冰水浴下将6.7kg的3,3-二甲基-1-氯-2-丁酮与1.4kg的1,2,4-三氮唑混合于冰冷的无水乙醇(500ml)中,加入碳酸钾6.9kg。混合均匀后,撤除冰水浴,使体系缓慢升温至室温,搅拌反应2小时。反应完毕,减压回收乙醇溶剂,水洗反应物,甲苯萃取三次,浓缩甲苯,使产品重结晶析出。过滤即得产物i,产品重量8250g,产率98%。
[0055]
冰水浴下,将8200g化合物i与7000g对氯苯甲醛溶于30l乙醇中,剧烈搅拌下将反应液搅拌冷却至0℃。随后,将2000g naoh溶于10l水中,并通过滴液漏斗将该naoh的水溶液缓慢滴加到冷却到0度的乙醇溶液中,控制滴加的温度为0-5℃。滴加完毕,撤除冰水浴,将反应液缓慢升温至室温,继续搅拌反应8小时。反应完毕,浓缩回收乙醇溶剂,过滤收集黄色的固体产物。所得的固体产物重新溶解于无水甲醇中,浓缩重结晶,得到的(e/z)-1-(4-氯苯基)-4,4-二甲基-2-(4h-1,2,4-三氮唑-4-基)-1-戊烯-3-酮产物为黄色针状晶体,产品重量12.40kg,产率90%。将该(e/z)-烯唑酮针状晶体混合产物重新溶解于无水氯仿中,氮气保护,紫外光照射条件下,室温反应12小时,使(e/z)-烯唑酮全部转位转化为(e)-1-(4-氯苯基)-4,4-二甲基-2-(4h-1,2,4-三氮唑-4-基)-1-戊烯-3-酮,产率100%。
[0056]
氮气保护下,投入(s)-吡咯啉-2-基-1,1,-二对甲苯基甲醇盐酸盐(7.15kg)、
氯苯基)-4,4-二甲基-2-(4h-1,2,4-三氮唑-4-基)-1-戊烯-3-酮产物为黄色针状晶体,产品重量1240kg,产率90%。将该(e/z)-烯唑酮针状晶体混合产物重新溶解于无水氯仿中,氮气保护,紫外光照射条件下,室温反应12小时,使(e/z)-烯唑酮全部转位转化为(e)-1-(4-氯苯基)-4,4-二甲基-2-(4h-1,2,4-三氮唑-4-基)-1-戊烯-3-酮,产率100%。
[0064]
氮气保护下,投入(s)-吡咯啉-2-基-1,1,-二对甲苯基甲醇盐酸盐(715kg)、nabh4(85kg)、50l二氯乙烷,以及三氯化铁(390kg)作为路易斯酸催化剂,放入-20℃的冷阱中30min,在搅拌下,2h内将试管中的溶液温度逐渐由-20℃升至室温。之后放入50℃油浴中,投入(e)-1-(4-氯苯基)-4,4-二甲基-2-(4h-1,2,4-三氮唑-4-基)-1-戊烯-3-酮(650kg)和5l dce,tlc监测反应进度。50℃反应4-8h。停止反应,用加入2n hcl水溶液(10l)淬灭反应并继续搅拌2h。50℃分层,水层用dcm 5x 200l萃取,合并的有机相用饱和食盐水洗涤,无水硫酸钠干燥。浓缩溶液,爬小板分出产物点用于测试ee值。剩余样重结晶,得到产物iii,605kg,产率90%,ee 77%。
[0065]
如上所述,尽管参照特定的优选实施例已经表示和表述了本发明,但其不得解释为对本发明自身的限制。在不脱离所附权利要求定义的本发明的精神和范围前提下,可对其在形式上和细节上作出各种变化。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1