包括使用新的铱催化剂对映选择性氢化4-取代的1,2-二氢喹啉的方法与流程
包括使用新的铱催化剂对映选择性氢化4-取代的1,2-二氢喹啉的方法
1.本发明涉及一种制备光学活性的4-取代的1,2,3,4-四氢喹啉的方法,其包括在手性铱(p,n)-配体催化剂的存在下,对映选择性氢化相应的4-取代的1,2-二氢喹啉。
2.由ep 0 654 464已知,n-乙酰基四氢喹啉可通过重排反应转化为相应的4-氨基茚满衍生物。
3.4-氨基茚满衍生物是用于制备各种具有杀真菌活性的n-茚满基杂芳基羧酰胺的重要中间体(ep 0 654 464、wo 2011/162397、wo 2012/084812、wo 2015/197530)。
4.ep 3 103 789公开了一种通过将对映体混合物转化成d-酒石酸的非对映体盐来光学拆分1,1,3-三甲基-4-氨基茚满的方法。在分离并碱化非对映体盐后,获得(r)-和(s)-1,1,3-三甲基-4-氨基茚满。该参考文献还公开了一种外消旋不希望的对映体的方法,使得整个方法能够通过几个方法步骤将不希望的对映体转化成希望的对映体。(r)-1,1,3-三甲基-4-氨基茚满是用于制备吡唑羧酰胺杀真菌剂inpyrfluxam的重要中间体。
5.通过不对称合成制备n-茚满基杂芳基羧酰胺手性中间体的方法也是已知的。wo 2015/141564记载了一种制备光学活性的4-取代的1,2,3,4-四氢喹啉的方法,该方法包括在具有光学活性配体的过渡金属催化剂的存在下,使相应的4-取代的1,2-二氢喹啉氢化。4-取代的nh-二氢喹啉的不对称氢化以中等转化率(最高达62.6%)和对映选择性(最高达71.3%ee)进行,而n-乙酰基二氢喹啉的转化率(最高达14%)和对映选择性(最高达31%ee)甚至更低。
6.基于上述现有技术,本发明的目的是提供一种制备光学活性的4-取代的1,2,3,4-四氢喹啉的方法,该方法优于现有技术的方法。该方法应能够以高产率和高对映体纯度制备所需的对映体,而方法步骤和纯化步骤很少。
7.上述目的通过一种制备式(ia)或(ib)的化合物的方法来实现,
[0008][0009]
其中
[0010]
r1选自c
1-c
6-烷基、c
1-c
6-卤代烷基、c
1-c
6-烷氧基-c
1-c
6-烷基、c
3-c
6-环烷基、c
6-c
14-芳基或c
6-c
14-芳基-c
1-c
4-烷基,
[0011]
其中c
1-c
6-烷基、c
3-c6环烷基和c
1-c
6-烷氧基-c
1-c
6-烷基部分中的c
1-c
6-烷氧基任选地被1至3个独立地选自卤素、c
1-c
4-烷氧基、c
1-c
4-卤代烷基、c
1-c
4-卤代烷氧基和苯基的取代基取代,其中苯基可被1至5个彼此独立地选自卤素、c
1-c
4-烷基、c
1-c
4-烷氧基、c
1-c
4-卤代烷基和c
1-c
4-卤代烷氧基的取代基取代,和
[0012]
其中c
6-c
14-芳基和c
6-c
14-芳基-c
1-c
4-烷基部分中的c
6-c
14-芳基在每种情况下为未取代的或被1至5个选自卤素、c
1-c
4-烷基、c
1-c
4-卤代烷基、c
1-c
4-烷氧基和c
1-c
4-卤代烷
氧基的取代基取代,
[0013]
r2和r3是相同的,并且选自氢、c
1-c
6-烷基、c
1-c
6-卤代烷基和c
1-c
6-烷氧基-c
1-c
6-烷基,
[0014]
或
[0015]
r2和r3与它们所键合的碳一起形成c
3-c
6-环烷基环,
[0016]
r4为氢、c
1-c
6-烷基、c
1-c
6-卤代烷基、c
1-c
6-烷氧基、c
1-c
6-卤代烷氧基、c
1-c
6-烷基氨基、c
2-c
6-烯基、c
2-c
6-炔基、c
3-c
6-环烷基、c
3-c
6-环烷基-c
1-c
4-烷基、c
2-c
6-烯氧基、9-芴基亚甲基氧基、c
6-c
14-芳基、c
6-c
14-芳氧基、c
6-c
14-芳基-c
1-c
4-烷氧基或c
6-c
14-芳基-c
1-c
4-烷基,
[0017]
其中c
6-c
14-芳基本身或作为复合取代基的一部分为未取代的或被1至5个选自卤素、c
1-c
4-烷基、c
1-c
4-卤代烷基、c
1-c
4-烷氧基和c
1-c
4-卤代烷氧基的取代基取代,n为0、1、2、3或4,
[0018]
每个取代基r5——如果存在——独立地选自卤素、c
1-c
6-烷基、c
1-c
6-卤代烷基、c
1-c
6-烷氧基、羟基、氨基和-c(=o)-c
1-c
6-烷基,
[0019]
其包括在手性铱催化剂的存在下,使式(ii)的化合物对映选择性氢化,
[0020][0021]
其中取代基r1、r2、r3、r4、r5和整数n各自如式(ia)或(ib)的化合物所定义,
[0022]
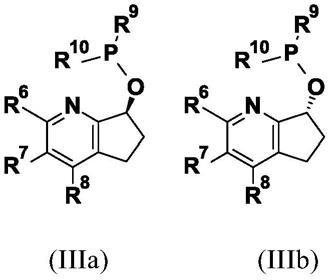
其特征在于手性铱催化剂包含式(iiia)或(iiib)的手性配体
[0023][0024]
其中
[0025]
r6是下式的基团
[0026][0027]
其中
[0028]
**表示与6,7-二氢-5h-环戊[b]吡啶部分连接的键,
[0029]r13
为氢、甲基或乙基,
[0030]r14
为c
1-c
6-烷基
[0031]
r7为氢,
[0032]
r8为c
1-c4烷基或苯基,其中苯基为未取代的或被1至5个选自卤素、c
1-c
4-烷基、c
1-c
4-卤代烷基、c
1-c
4-烷氧基和c
1-c
4-卤代烷氧基的取代基取代,
[0033]
r9和r
10
彼此独立地选自c
1-c
6-烷基、c
3-c
8-环烷基、哌啶基和吡啶基,
[0034]
或
[0035]
r9和r
10
与它们所键合的磷原子一起形成基团g1或g2[0036][0037]
其中由“x”和“y”标识的键都直接与磷原子键合,
[0038]
p和q彼此独立地选自0、1和2,
[0039]r11
和r
12
独立地选自c
1-c
6-烷基和苯基,其可被1至5个选自卤素、c
1-c
4-烷基、c
1-c
4-烷氧基和苯基的取代基取代,所述苯基取代基可被一个或两个c
1-c
4-烷基取代基取代,
[0040]
或
[0041]
r9和r
10
与它们所键合的磷原子一起形成g3基团
[0042][0043]
其中由“u”和“v”标识的键都直接与磷原子键合。
[0044]
出乎意料地发现,在手性铱(p,n)-配体催化剂的存在下,光学活性的4-取代的1,2,3,4-四氢喹啉(ia和ib)可通过使相应的4-取代的1,2-二氢喹啉(ii)对映选择性氢化而以高产率和优异的对映选择性制备。
[0045]
定义
[0046]
在上式给出的符号定义中,使用通常表示以下取代基的集合术语:
[0047]
卤素:氟、氯、溴或碘,优选氟、氯或溴,更优选氟或氯。
[0048]
烷基:具有1至6个、优选1至4个碳原子的饱和直链或支链的烃基取代基,例如(但不限于)c
1-c
6-烷基,例如甲基、乙基、丙基(正丙基)、1-甲基乙基(异丙基)、丁基(正丁基)、1-甲基丙基(仲丁基)、2-甲基丙基(异丁基)、1,1-二甲基乙基(叔丁基)、戊基、1-甲基丁基、2-甲基丁基、3-甲基丁基、2,2-二甲基丙基、1-乙基丙基、1,1-二甲基丙基、1,2-二甲基丙
基、己基、1-甲基戊基、2-甲基戊基、3-甲基戊基、4-甲基戊基、1,1-二甲基丁基、1,2-二甲基丁基、1,3-二甲基丁基、2,2-二甲基丁基、2,3-二甲基丁基、3,3-二甲基丁基、1-乙基丁基、2-乙基丁基、1,1,2-三甲基丙基、1,2,2-三甲基丙基、1-乙基-1-甲基丙基和1-乙基-2-甲基丙基。特别地,所述基团为c
1-c
4-烷基,如甲基、乙基、丙基、1-甲基乙基(异丙基)、丁基、1-甲基丙基(仲丁基)、2-甲基丙基(异丁基)或1,1-二甲基乙基(叔丁基)。除非另有定义,否则此定义还适用于作为复合取代基的一部分的烷基,例如c
3-c6环烷基-c
1-c
4-烷基、c
6-c
14-芳基-c
1-c
4-烷基等。
[0049]
烯基:具有2至6个、优选2至4个碳原子和一个在任何位置处的双键的不饱和直链或支链的烃基取代基,例如(但不限于)c
2-c
6-烯基,例如乙烯基、烯丙基、(e)-2-甲基乙烯基、(z)-2-甲基乙烯基、异丙烯基、高烯丙基、(e)-丁-2-烯基、(z)-丁-2-烯基、(e)-丁-1-烯基、(z)-丁-1-烯基、2-甲基丙-2-烯基、1-甲基丙-2-烯基、2-甲基丙-1-烯基、(e)-1-甲基丙-1-烯基、(z)-1-甲基丙-1-烯基、戊-4-烯基、(e)-戊-3-烯基、(z)-戊-3-烯基、(e)-戊-2-烯基、(z)-戊-2-烯基、(e)-戊-1-烯基、(z)-戊-1-烯基、3-甲基丁-3-烯基、2-甲基丁-3-烯基、1-甲基丁-3-烯基、3-甲基丁-2-烯基、(e)-2-甲基丁-2-烯基、(z)-2-甲基丁-2-烯基、(e)-1-甲基丁-2-烯基、(z)-1-甲基丁-2-烯基、(e)-3-甲基丁-1-烯基、(z)-3-甲基丁-1-烯基、(e)-2-甲基丁-1-烯基、(z)-2-甲基丁-1-烯基、(e)-1-甲基丁-1-烯基、(z)-1-甲基丁-1-烯基、1,1-二甲基丙-2-烯基、1-乙基丙-1-烯基、1-丙基乙烯基、1-异丙基乙烯基、(e)-3,3-二甲基丙-1-烯基、(z)-3,3-二甲基丙-1-烯基、己-5-烯基、(e)-己-4-烯基、(z)-己-4-烯基、(e)-己-3-烯基、(z)-己-3-烯基、(e)-己-2-烯基、(z)-己-2-烯基、(e)-己-1-烯基、(z)-己-1-烯基、4-甲基戊-4-烯基、3-甲基戊-4-烯基、2-甲基戊-4-烯基、1-甲基戊-4-烯基、4-甲基戊-3-烯基、(e)-3-甲基戊-3-烯基、(z)-3-甲基戊-3-烯基、(e)-2-甲基戊-3-烯基、(z)-2-甲基戊-3-烯基、(e)-1-甲基戊-3-烯基、(z)-1-甲基戊-3-烯基、(e)-4-甲基戊-2-烯基、(z)-4-甲基戊-2-烯基、(e)-3-甲基戊-2-烯基、(z)-3-甲基戊-2-烯基、(e)-2-甲基戊-2-烯基、(z)-2-甲基戊-2-烯基、(e)-1-甲基戊-2-烯基、(z)-1-甲基戊-2-烯基、(e)-4-甲基戊-1-烯基、(z)-4-甲基戊-1-烯基、(e)-3-甲基戊-1-烯基、(z)-3-甲基戊-1-烯基、(e)-2-甲基戊-1-烯基、(z)-2-甲基戊-1-烯基、(e)-1-甲基戊-1-烯基、(z)-1-甲基戊-1-烯基、3-乙基丁-3-烯基、2-乙基丁-3-烯基、1-乙基丁-3-烯基、(e)-3-乙基丁-2-烯基、(z)-3-乙基丁-2-烯基、(e)-2-乙基丁-2-烯基、(z)-2-乙基丁-2-烯基、(e)-1-乙基丁-2-烯基、(z)-1-乙基丁-2-烯基、(e)-3-乙基丁-1-烯基、(z)-3-乙基丁-1-烯基、2-乙基丁-1-烯基、(e)-1-乙基丁-1-烯基、(z)-1-乙基丁-1-烯基、2-丙基丙-2-烯基、1-丙基丙-2-烯基、2-异丙基丙-2-烯基、1-异丙基丙-2-烯基、(e)-2-丙基丙-1-烯基、(z)-2-丙基丙-1-烯基、(e)-1-丙基丙-1-烯基、(z)-1-丙基丙-1-烯基、(e)-2-异丙基丙-1-烯基、(z)-2-异丙基丙-1-烯基、(e)-1-异丙基丙-1-烯基、(z)-1-异丙基丙-1-烯基、1-(1,1-二甲基乙基)乙烯基、丁-1,3-二烯基、戊-1,4-二烯基、己-1,5-二烯基或甲基己二烯基。特别地,所述基团为乙烯基或烯丙基。除非另有定义,否则此定义还适用于作为复合取代基的一部分的烯基。
[0050]
炔基:具有2至8个、优选2至6个、更优选2至4个碳原子和一个在任何位置处的三键的直链或支链烃基取代基,例如(但不限于)c
2-c
6-炔基,例如乙炔基、丙-1-炔基、丙-2-炔基、丁-1-炔基、丁-2-炔基、丁-3-炔基、1-甲基丙-2-炔基、戊-1-炔基、戊-2-炔基、戊-3-炔基、戊-4-炔基、2-甲基丁-3-炔基、1-甲基丁-3-炔基、1-甲基丁-2-炔基、3-甲基丁-1-炔基、
1-乙基丙-2-炔基、己-1-炔基、己-2-炔基、己-3-炔基、己-4-炔基、己-5-炔基、3-甲基戊-4-炔基、2-甲基戊-4-炔基、1-甲基戊-4-炔基、2-甲基戊-3-炔基、1-甲基戊-3-炔基、4-甲基戊-2-炔基、1-甲基戊-2-炔基、4-甲基戊-1-炔基、3-甲基戊-1-炔基、2-乙基丁-3-炔基、1-乙基丁-3-炔基、1-乙基丁-2-炔基、1-丙基丙-2-炔基、1-异丙基丙-2-炔基、2,2-二甲基丁-3-炔基、1,1-二甲基丁-3-炔基、1,1-二甲基丁-2-炔基或3,3-二甲基丁-1-炔基。特别地,所述炔基为乙炔基、丙-1-炔基或丙-2-炔基。除非另有定义,否则此定义还适用于作为复合取代基的一部分的炔基。
[0051]
烷基氨基:单烷基氨基或二烷基氨基,其中单烷基氨基表示具有一个烷基残基的氨基,所述烷基残基具有1至4个碳原子且与氮原子连接。非限制性实例包括甲基氨基、乙基氨基、正丙基氨基、异丙基氨基、正丁基氨基和叔丁基氨基。其中二烷基氨基表示具有两个独立选择的烷基残基的氨基,所述烷基残基具有1至4个碳原子且各自与氮原子连接。非限制性实例包括n,n-二甲基氨基、n,n-二乙基氨基、n,n-二异丙基氨基、n-乙基-n-甲基氨基、n-甲基-n-正丙基氨基、n-异丙基-n-正丙基氨基和n-叔丁基-n-甲基氨基。
[0052]
烷氧基:具有1至6个、更优选1至4个碳原子的饱和直链或支链烷氧基取代基,例如(但不限于)c
1-c
6-烷氧基,例如甲氧基、乙氧基、丙氧基、1-甲基乙氧基、丁氧基、1-甲基丙氧基、2-甲基丙氧基、1,1-二甲基乙氧基、戊氧基、1-甲基丁氧基、2-甲基丁氧基、3-甲基丁氧基、2,2-二甲基丙氧基、1-乙基丙氧基、1,1-二甲基丙氧基、1,2-二甲基丙氧基、己氧基、1-甲基戊氧基、2-甲基戊氧基、3-甲基戊氧基、4-甲基戊氧基、1,1-二甲基丁氧基、1,2-二甲基丁氧基、1,3-二甲基丁氧基、2,2-二甲基丁氧基、2,3-二甲基丁氧基、3,3-二甲基丁氧基、1-乙基丁氧基、2-乙基丁氧基、1,1,2-三甲基丙氧基、1,2,2-三甲基丙氧基、1-乙基-1-甲基丙氧基和1-乙基-2-甲基丙氧基。除非另有定义,否则此定义还适用于作为复合取代基的一部分的烷氧基。
[0053]
环烷基:具有3至12个、优选3至8个、更优选3至6个碳环成员的单环或多环饱和烃基取代基,例如(但不限于)环丙基、环戊基、环己基和金刚烷基。除非另有定义,此定义还适用于作为复合取代基的一部分的环烷基,例如c
3-c
6-环烷基-c
1-c
4-烷基。
[0054]
卤代烷基:具有1至6个、优选1至4个碳原子的直链或支链烷基取代基(如上所述),其中这些基团中的一些或全部氢原子被如上所述的卤素原子取代,例如(但不限于)c
1-c
3-卤代烷基,例如氯甲基、溴甲基、二氯甲基、三氯甲基、氟甲基、二氟甲基、三氟甲基、氯氟甲基、二氯氟甲基、氯二氟甲基、1-氯乙基、1-溴乙基、1-氟乙基、2-氟乙基、2,2-二氟乙基、2,2,2-三氟乙基、2-氯-2-氟乙基、2-氯-2,2-二氟乙基、2,2-二氯-2-氟乙基、2,2,2-三氯乙基、五氟乙基和1,1,1-三氟丙-2-基。除非另有定义,此定义还适用于作为复合取代基的一部分的卤代烷基。
[0055]
卤代烯基和卤代炔基类似于卤代烷基定义,不同之处在于烯基和炔基而不是烷基作为取代基的一部分存在。
[0056]
卤代烷氧基:具有1至6个、优选1至4个碳原子的直链或支链烷氧基取代基(如上所述),其中这些基团中的一些或全部氢原子被如上所述的卤素原子取代,例如(但不限于)c
1-c
3-卤代烷氧基,例如氯甲氧基、溴甲氧基、二氯甲氧基、三氯甲氧基、氟甲氧基、二氟甲氧基、三氟甲氧基、氯氟甲氧基、二氯氟甲氧基、氯二氟甲氧基、1-氯乙氧基、1-溴乙氧基、1-氟乙氧基、2-氟乙氧基、2,2-二氟乙氧基、2,2,2-三氟乙氧基、2-氯-2-氟乙氧基、2-氯-2,2-二
氟乙氧基、2,2-二氯-2-氟乙氧基、2,2,2-三氯乙氧基、五氟乙氧基和1,1,1-三氟丙-2-氧基。除非另有定义,此定义还适用于作为复合取代基的一部分的卤代烷氧基。
[0057]
芳基:具有6至14个碳原子的单环、双环或三环芳族或部分芳族取代基,例如(但不限于)苯基、萘基、四氢萘基、茚基和茚满基。可通过芳基残基的任一可能的环成员与上级通用结构键合。芳基优选选自苯基、1-萘基、2-萘基、9-菲基和9-蒽基。尤其优选苯基。
[0058]
如本文所用,术语“对映选择性”意指优选形成氢化产物的两种可能的对映体中的一种,即式(ia)的对映体或式(ib)的对映体。“对映体过量”或“ee”表示对映选择性的程度:
[0059][0060]
通过选择手性配体,例如通过选择式(iiia)的手性配体或相反的对映体(式(iiib)的配体),可控制主要对映体。
[0061]
本发明的方法用于制备式(ia)或(ib)、优选式(ia)的化合物。
[0062]
优选式(ia)或(ib)、尤其是式(ia)的化合物,其中取代基定义如下:
[0063]
r1为c
1-c
6-烷基或c
6-c
14-芳基-c
1-c
4-烷基,
[0064]
其中c
6-c
14-芳基-c
1-c
4-烷基部分中的c
6-c
14-芳基是未取代的或被1至5个选自卤素、c
1-c
4-烷基、c
1-c
4-卤代烷基、c
1-c
4-烷氧基和c
1-c
4-卤代烷氧基的取代基取代,
[0065]
r2和r3是相同的,并且选自c
1-c
4-烷基,
[0066]
r4为c
1-c
4-烷基、c
1-c
4-卤代烷基、c
1-c
4-烷氧基、c
1-c
4-卤代烷氧基、苯基或苄基,
[0067]
n为0、1或2,
[0068]
每个取代基r5——如果存在——独立地选自卤素、c
1-c
6-烷基和c
1-c6卤代烷基。
[0069]
更优选式(ia)或(ib)、尤其是式(ia)的化合物,其中取代基定义如下:
[0070]
r1为c
1-c
6-烷基,
[0071]
r2和r3是相同的,并且选自c
1-c
4-烷基,
[0072]
或
[0073]
r2和r3连同它们所键合的碳一起形成c
3-c
6-环烷基环,
[0074]
r4为c
1-c
4-烷基、c
1-c
4-卤代烷基、苯基或苄基,
[0075]
n为0、1或2,
[0076]
每个取代基r5——如果存在——独立地选自卤素和c
1-c
6-烷基。
[0077]
甚至更优选式(ia)或(ib)、尤其是(ia)的化合物,其中取代基定义如下:
[0078]
r1为甲基、乙基或正丙基,
[0079]
r2和r3为甲基,
[0080]
r4为c
1-c
4-烷基,
[0081]
n为0、1或2,
[0082]
每个取代基r5——如果存在——独立地选自卤素和c
1-c
6-烷基。
[0083]
最优选式(ia)或(ib)、尤其是(ia)的化合物,其中取代基定义如下:
[0084]
r1为甲基或正丙基,
[0085]
r2和r3为甲基,
[0086]
r4为甲基,
[0087]
n为0或1,
[0088]
取代基r5——如果存在——为氟。
[0089]
本发明的方法包括对映选择性氢化式(ii)的化合物。式(ii)的化合物中的取代基r1、r2、r3、r4、r5和整数n各自如式(ia)或(ib)的化合物所定义。
[0090]
对映选择性氢化式(ii)的化合物在包含式(iiia)或(iiib)的手性配体的手性铱催化剂的存在下进行。
[0091]
在本发明方法的一个优选实施方案中,式(ia)、(ib)、(ii)、(iiia)、(iiib)的取代基定义如下:
[0092]
r1为c
1-c
6-烷基或c
6-c
14-芳基-c
1-c
4-烷基,
[0093]
其中c
6-c
14-芳基-c
1-c
4-烷基部分中的c
6-c
14-芳基为未取代的或被1至5个选自卤素、c
1-c
4-烷基、c
1-c
4-卤代烷基、c
1-c
4-烷氧基和c
1-c
4-卤代烷氧基的取代基取代
[0094]
r2和r3相同并且选自c
1-c
4-烷基,
[0095]
r4为c
1-c
4-烷基、c
1-c
4-卤代烷基、c
1-c
4-烷氧基、c
1-c
4-卤代烷氧基、苯基或苄基,
[0096]
n为0、1或2,
[0097]
每个取代基r5——如果存在——独立地选自卤素、c
1-c
6-烷基和c
1-c
6-卤代烷基,
[0098]
r6为下式的基团
[0099][0100]
其中
[0101]
**表示与6,7-二氢-5h-环戊[b]吡啶部分连接的键,
[0102]r13
为氢、甲基或乙基,
[0103]r14
为c
1-c4烷基
[0104]
r7为氢,
[0105]
r8为c
1-c4烷基或苯基,
[0106]
其中苯基为未取代的或被一至五个c
1-c
4-烷基取代基取代,r9和r
10
彼此独立地选自异丙基、叔丁基、环戊基、环己基和哌啶-1-基。
[0107]
在本发明方法的一个更优选的实施方案中,式(ia)、(ib)、(ii)、(iiia)、(iiib)的取代基定义如下:
[0108]
r1为c
1-c
6-烷基,
[0109]
r2和r3相同并且选自c
1-c
4-烷基,
[0110]
r4为c
1-c
4-烷基、c
1-c
4-卤代烷基、c
1-c
4-烷氧基、c
1-c
4-卤代烷氧基、苯基或苄基,
[0111]
n为0、1或2,
[0112]
每个取代基r5——如果存在——独立地选自卤素、c
1-c
6-烷基和c
1-c
6-卤代烷基,
[0113]
r6为下式的基团,
[0114][0115]
其中
[0116]
**表示与6,7-二氢-5h-环戊[b]吡啶部分连接的键,
[0117]r13
为乙基,
[0118]r14
为甲基,
[0119]
r7为氢,
[0120]
r8为甲基,
[0121]
r9和r
10
彼此独立地选自环己基和哌啶-1-基。
[0122]
在本发明方法的最优选的实施方案中,式(ia)、(ib)、(ii)、(iiia)、(iiib)的取代基定义如下:
[0123]
r1为c
1-c
4-烷基,
[0124]
r2和r3为甲基,
[0125]
r4为c
1-c
4-烷基,
[0126]
n为0或1
[0127]
r5如果存在,为氟,
[0128]
r6为2,6-二乙基-4-甲基苯基,
[0129]
r7为氢,
[0130]
r8为甲基
[0131]
r9和r
10
均为环己基。
[0132]
取决于是化合物(ia)还是(ib)为所需的产物,对式(iiia)或(iiib)的配体进行选择。
[0133]
优选式(iiia)和(iiib)的配体,其中取代基定义如下:
[0134]
r6为2,6-二乙基-4-甲基苯基,
[0135]
r7为氢,
[0136]
r8为甲基,
[0137]
r9和r
10
均为环己基
[0138]
优选地,手性铱催化剂选自[irl*(cod)]y和[irl*(nbd)]y,其中
[0139]
l*为式(iiia)和(iiib)的手性配体,
[0140]
cod表示1,5-环辛二烯,
[0141]
nbd表示降冰片二烯,并且
[0142]
y为选自[b(r
18
)4]-、pf
6-和式(vii)的[al{oc(cf3)3}4]-的非配位阴离子
[0143][0144]
其中r
18
选自氟和苯基,其为未取代的或被1至5个选自c
1-c
4-烷基、c
1-c
4-卤代烷基和卤素的取代基取代。
[0145]
更优选式[irl*(cod)]y和[irl*(nbd)]y的手性铱催化剂,其中y为式(vii)的[al{oc(cf3)3}4]-或[b(r
18
)4]-,其中r
18
为苯基,其为未取代的或被1至5个选自氟和三氟甲基的取代基取代。
[0146]
甚至更优选通式(va)和(vb)的手性铱催化剂
[0147][0148]
其中
[0149]
r6为2,6-二乙基-4-甲基苯基,
[0150]
r7为氢,
[0151]
r8为甲基
[0152]
r9和r
10
均为环己基
[0153]
y为选自[b(r
18
)4]-和式(vii)的[al{oc(cf3)3}4]-的非配位阴离子,
[0154]
其中r
18
为3,5-双(三氟甲基)苯基。
[0155]
最优选的是通式(va)的手性铱催化剂
[0156][0157]
其中
[0158]
r6为2,4,6-三甲基苯基,
[0159]
r7为氢,
[0160]
r8为甲基
[0161]
r9和r
10
均为环己基
[0162]
y为式(vii)的[al{oc(cf3)3}4]-。
[0163]
铱催化剂的用量优选为0.001mol%至5mol%、更优选为0.001mol%至4mol%、最优选为0.002mol%至3mol%、尤其是0.005mol%至1.0mol%,基于式(ii)的化合物的量计。
[0164]
手性铱催化剂可通过本领域已知的方法由铱(i)催化剂前体(例如[ir(cod)cl]2)、式(iiia)或(iiib)的手性配体和非配位阴离子的碱金属盐制备(s.kaiser等人,angew.chem.int.ed.2006,45,5194-5197;w.j.druryiii等人,angew.chem.int.ed.2004,43,70-74)。
[0165]
本发明的方法包括使式(ii)的化合物对映选择性氢化。
[0166]
优选地,氢化使用氢气在1至300巴、优选3至200巴、最优选20至150巴的压力下进行。
[0167]
氢化优选在20℃至130℃、更优选30℃至100℃的温度下进行。
[0168]
合适的溶剂为卤代醇例如2,2,2-三氟乙醇、六氟异丙醇(1,1,1,3,3,3-六氟-2-丙醇)和四氟丙醇(2,2,3,3-四氟-1-丙醇),卤代烃例如氯苯、二氯苯、二氯甲烷、氯仿、四氯甲烷、二氯乙烷和三氯乙烷,芳族烃例如苯、甲苯和二甲苯,醚例如乙醚、二异丙醚、甲基叔丁基醚、甲基叔戊基醚、二噁烷、四氢呋喃、1,2-二甲氧基乙烷、1,2-二乙氧基乙烷和苯甲醚,以及酯例如乙酸乙酯、乙酸异丙酯,及其混合物。
[0169]
优选的溶剂选自2,2,2-三氟乙醇、六氟异丙醇、1,2-二氯乙烷、四氟丙醇、1,4-二噁烷、乙酸异丙酯、甲苯及其混合物。
[0170]
更优选的溶剂选自2,2,2-三氟乙醇、六氟异丙醇、1,2-二氯乙烷、四氟丙醇及其混合物。
[0171]
特别优选2,2,2-三氟乙醇和六氟异丙醇。
[0172]
最优选六氟异丙醇。
[0173]
本发明的方法可任选地在添加剂的存在下进行,所述添加剂选自布朗斯特酸和路易斯酸。
[0174]
在本发明的方法的优选实施方案中,添加剂选自六氟磷酸、乙酸、三氟甲基磺酸、
水、五氟苯酚、3,5-双(三氟甲基)苯酚、四氟硼酸、四氟硼酸二乙醚配合物、全氟磺酸基聚合物(nafion)、amberlyst、1,1,1,3,3,3-六氟-2-(三氟甲基)丙-2-醇、三苯基硼烷、三[3,5-双(三氟甲基)苯基]硼烷、三(2,3,4,5,6-五氟苯基)硼烷、硼烷四氢呋喃配合物、硼酸、三氟甲烷磺酸铝(iii)、三氟甲烷磺酸锌(ii)、三氟甲烷磺酸钪(iii)、氟化铝(iii)、异丙醇钛(iv)、三甲基铝、三氟化硼、三氟化硼配合物及其混合物。
[0175]
合适的三氟化硼配合物为三氟化硼与有机溶剂(例如二烷基醚或醇)的配合物,以及三氟化硼与有机酸(例如羧酸)的配合物。优选的三氟化硼配合物选自三氟化硼二乙醚配合物、三氟化硼乙酸配合物和三氟化硼正丙醇配合物。
[0176]
在本发明的方法的更优选实施方案中,添加剂选自六氟磷酸、五氟苯酚、3,5-双(三氟甲基)苯酚、四氟硼酸二乙醚配合物、三苯基硼烷、三[3,5-双(三氟甲基)苯基]硼烷、三(2,3,4,5,6-五氟苯基)硼烷、三氟甲烷磺酸铝(iii)、三氟甲烷磺酸钪(iii)、氟化铝(iii)、异丙醇钛(iv)、三甲基铝、三氟化硼、三氟化硼配合物及其混合物,其中三氟化硼配合物优选选自三氟化硼二乙醚配合物、三氟化硼乙酸配合物和三氟化硼正丙醇配合物。
[0177]
在本发明的方法的甚至更优选实施方案中,添加剂选自六氟磷酸、五氟苯酚、3,5-双(三氟甲基)苯酚、三苯基硼烷、三[3,5-双(三氟甲基)苯基]硼烷、三(2,3,4,5,6-五氟苯基)硼烷、三氟甲烷磺酸铝(iii)、三氟甲烷磺酸钪(iii)、氟化铝(iii)、异丙醇钛(iv)、三甲基铝、三氟化硼、三氟化硼配合物及其混合物,其中三氟化硼配合物优选选自三氟化硼二乙醚配合物、三氟化硼乙酸配合物和三氟化硼正丙醇配合物。
[0178]
在本发明的方法的最优选实施方案中,添加剂最优选选自三氟甲烷磺酸铝(iii)、三氟甲烷磺酸钪(iii)、三(2,3,4,5,6-五氟苯基)硼烷、六氟磷酸、三氟化硼和三氟化硼配合物,其中三氟化硼配合物优选选自三氟化硼二乙醚配合物、三氟化硼乙酸配合物和三氟化硼正丙醇配合物。
[0179]
选自布朗斯特酸和路易斯酸的添加剂的用量优选为0.1mol%至10mol%、更优选为0.2mol%至5mol%、最优选为0.3mol%至2mol%、尤其是0.4mol%至1mol%,基于式(ii)的化合物的量计。
[0180]
缩略语和首字母缩写:
[0181][0182][0183]
制备铱催化剂
[0184][0185]
配体前体(富含对映体的仲醇)根据已知的文献方法类似于以下文献中公开的方法进行制备:s.kaiser等人,angew.chem.int.ed.2006,45,5194-5197或d.h.woodmansee chem.sci 2010,1,72。配体和铱配合物基于相同的文献先例通过改造的方法进行制备:
[0186]
标准方法
[0187]
配体合成方法(在ar下):将醇前体的thf溶液(0.25mmol,于5.0ml thf)冷却至-78℃,并将n-buli(0.1ml 2.5m n-buli的己烷溶液;0.25mmol;1当量)逐滴添加至持续搅拌的溶液中。添加完成后,使溶液温热至室温,并在此温度下搅拌另外30分钟。将溶液再次冷却
至-78℃,并将r9r
10
pcl(0.25mmol,1当量)添加至持续搅拌的溶液中。使混合物温热至室温,随后加热至50℃,并在此温度保持过夜。使用
31
p-nmr计算配体的理论产率,并将该配体无需进一步纯化即用于下一步骤。
[0188]
配合方法(在ar下):向粗配体溶液中添加[ir(cod)2]barf(barf=四[3,5-双(三氟甲基)苯基]-硼酸根)(呈固体,1当量,基于理论产率计)。将得到的混合物加热至50℃,并在此温度下保持3小时。
[0189]
后处理(在空气下):冷却至室温后,将反应溶液旋转蒸发至二氧化硅上,负载于二氧化硅柱上。使用戊烷/乙醚洗脱副组分,随后用dcm洗脱所需的配合物。然后在减压下蒸发溶剂。
[0190]
合成并表征以下所示的催化剂:
[0191][0192]
表1:
[0193]
催化剂r8r9,r
10
r7r
13r14
[y]-va-1甲基环己基hetme四(3,5-双(三氟甲基)苯基)硼酸根va-2甲基环己基hetme式(vii)的[al{oc(cf3)3}
4-va-3苯基环己基hetme四(3,5-双(三氟甲基)苯基)硼酸根va-4甲基s-binol*-基hetme四(3,5-双(三氟甲基)苯基)硼酸根va-5甲基哌啶-1-基hetme四(3,5-双(三氟甲基)苯基)硼酸根va-6甲基环己基hmetbupf
6-va-7甲基cg-phos**hmeh四(3,5-双(三氟甲基)苯基)硼酸根va-8甲基叔丁基,2-吡啶基hmeh四(3,5-双(三氟甲基)苯基)硼酸根va-9甲基环己基hmeh式(vii)的[al{oc(cf3)3}4]-va-10甲基环己基hmeme四(3,5-双(三氟甲基)苯基)硼酸根va-11甲基环己基hmetbu四(3,5-双(三氟甲基)苯基)硼酸根va-12甲基环己基hmeh四(3,5-双(三氟甲基)苯基)硼酸根va-13h苯基hhh四(3,5-双(三氟甲基)苯基)硼酸根
[0194]
binol*(s)-(-)-1,1
′‑
二萘-2,2
′‑
二醇
[0195]
[0196]
va-1
[0197]
根据上述标准方法进行反应。配合物可以橙色固体形式分离(282mg;76%基于[ir(cod)2]barf)。
[0198]1h-nmr(300mhz,cd2cl2):δ(ppm)=7.73(s,8h),7.56(s,4h),7.23(s,1h),7.14(s,1h),7.00(s,1h),5.62(dd,j=8.0,5.6hz,1h),5.46
–
5.39(m,1h),4.32(dd,j=7.4,3.4hz,1h),3.36
–
3.27(m,1h),3.19
–
3.06(m,2h),3.01
–
2.91(m,2h),2.80(dq,j=14.9,7.4hz,1h),2.59
–
2.43(m,2h),2.43
–
2.15(m,7h),2.15
–
0.83(m,36h),0.66
–
0.48(m,1h)。
31
p-nmr(122mhz,cd2cl2)δ(ppm)=119.00。
19
f-nmr(282mhz,cd2cl2)δ(ppm)=-62.87。hr-ms(esi)m/z c
40h58
nopir[m]
+
计算值792.3880,实测值792.3903。
[0199]
va-2
[0200]
将相应的醇前体的thf溶液(0.25mmol,于5.0ml thf中)冷却至-78℃,并将n-buli(0.1ml 2.5m n-buli的己烷溶液;0.25mmol;1当量)滴加到连续搅拌的溶液中。加入完成后,使溶液温热至室温,并在该温度下再搅拌30分钟。将溶液再次冷却至-78℃,并将cy2pcl(0.25mmol,1当量)加入到连续搅拌的溶液中。使混合物温热至室温,随后加热至50℃,并在该温度下保持过夜。在反应冷却至室温后,除去thf并在真空中干燥,将[ir(cod)cl]2(0.125mmol)和dcm(5.0ml)加入到试管中,在50℃下搅拌2小时。然后将li{al[oc(cf3)3]4}(0.275mmol)加入到反应混合物中并在室温下搅拌过夜。将反应溶液旋转蒸发到二氧化硅上,负载于用dcm制备的二氧化硅柱上,用正庚烷/dcm:1/1色谱分离,得到橙色固体(140mg,32%)。
[0201]1h-nmr(300mhz,cd2cl2)δ=7.27(s,1h),7.18(s,1h),7.04(s,1h),5.66(dt,j=8.0,3.8hz,1h),5.50-5.44(m,1h),4.40-4.31(m,1h),3.46-3.26(m,1h),3.25-2.91(m,4h),2.91-2.77(m,1h),2.57-2.52(m,2h),2.50-2.20(m,7h),2.19-1.78(m,13h),1.70-1.53(m,5h),1.49-1.02(m,18h),0.62(s,1h)。
31
p-nmr(122mhz,cd2cl2)δ=119.02。
19
f-nmr(282mhz,cd2cl2)δ=-75.74。hr-ms(esi)m/z c
45h60
nopir[m]
+
计算值792.3880,实测值792.3903。
[0202]
va-3
[0203]
根据上述标准方法进行反应。配合物可以橙色固体形式分离(162mg;42%基于[ir(cod)2]barf)。
[0204]1h-nmr(300mhz,cd2cl2)δ=7.80-7.65(m,8h),7.65-7.41(m,10h),7.17(s,1h),7.04(s,1h),5.70-5.65(m,1h),5.53-5.47(m,1h),4.42-4.35(m,1h),3.43-3.32(m,2h),3.27-3.09(m,2h),3.09-2.93(m,1h),2.86(dq,j=14.8,7.4hz,1h),2.55-2.48(m,2h),2.38(s,3h),2.27-1.46(m,19h),1.46-0.74(m,18h),0.69-0.52(m,1h)。
31
p-nmr(122mhz,cd2cl2)δ=119.07。
19
f-nmr(282mhz,cd2cl2)δ=-62.87。hr-ms(esi)m/z c
45h60
nopir[m]
+
计算值854.4036,实测值854.4073。
[0205]
va-4
[0206]
根据上述标准方法,使用clp-(s)-binol(0.25mmol,新鲜制备的)和[ir(cod)2]barf(287mg,0.225mmol)进行反应。在使用庚烷/dcm:1/1进行三次柱层析后,配合物可以橙色固体形式分离(104mg)。
31
p nmr中仍显示有四个峰,主峰为117.72ppm(纯度约为80%)。hrms显示存在所期望的配合物。
[0207]
31
p-nmr(122mhz,cd2cl2)δ=120.36,117.72,114.61,92.27。
19
f-nmr(282mhz,cd2cl2)δ=-62.85。hr-ms(esi)m/z c
48h48
no3pir[m]
+
计算值910.2996,实测值910.3026。
[0208]
va-5
[0209]
根据上述标准方法,使用1,1'-(氯磷烷二基)二哌啶(0.25mmol,新制备的)进行反应。配合物可以红色固体形式分离(149mg)。
31
p nmr显示6个峰,主峰在102.35和98.87ppm(2:3)处。hrms显示存在所期望的配合物。
[0210]
31
p-nmr(122mhz,cd2cl2)δ=102.35,101.96,101.40,98.87,98.46,98.35。
19
f-nmr(282mhz,cd2cl2)δ=-62.87。hr-ms(esi)m/z c
39h58
n3opir[m]
+
计算值808.3941,实测值808.3958。
[0211]
va-6
[0212]
将相应的醇前体的thf溶液(0.25mmol,于5.0ml thf中)冷却至-78℃,并将n-buli(0.1ml 2.5m n-buli的己烷溶液;0.25mmol;1当量)逐滴添加至持续搅拌的溶液中。添加完成后,使溶液温热至室温,并在此温度下搅拌另外30分钟。将溶液再次冷却至-78℃,并将cy2pcl(0.25mmol,1当量)添加至持续搅拌的溶液中。使混合物温热至室温,随后加热至50℃,并在此温度下保持过夜。反应冷却至室温后,除去thf并真空干燥,将[ir(cod)cl]2(0.125mmol)和dcm(5.0ml)添加至试管中,于50℃搅拌2小时。然后将kpf6(0.25mmol)添加至反应混合物,并在室温下搅拌过夜。将反应溶液旋转蒸发至二氧化硅上,负载于用dcm制成的二氧化硅柱上,用etoac/dcm:1/10进行层析,在两次柱层析后得到橙色固体(130mg,55%)。
[0213]1h-nmr(300mhz,cd2cl2)δ=7.37
–
7.02(m,3h),5.74
–
5.56(m,1h),5.52
–
5.46(m,1h),4.47
–
4.30(m,1h),3.45
–
3.21(m,1h),3.19
–
2.92(m,3h),2.66(s,3h),2.63
–
2.48(m,2h),2.44(s,3h),2.40
–
2.19(m,2h),2.16
–
1.70(m,15h),1.68
–
1.46(m,6h),1.41
–
1.28(m,13h),1.18
–
0.95(m,5h),0.71
–
0.58(m,1h)。
31
p-nmr(122mhz,cd2cl2)δ=118.42,-127.01,-132.85,-138.70,-144.55,-150.39,-156.24,-162.09。
19
f-nmr(376mhz,cd2cl2)δ=-72.64,-74.52。hr-ms(esi)m/z c
41h60
nopir[m]
+
计算值806.4036,实测值806.4061.
[0214]
va-7
[0215]
根据上述标准方法,使用cgpbr(0.25mmol,新鲜制备的)进行反应。配合物可以橙色固体形式分离(120mg)。在
31
p nmr中显示两个峰。hrms显示存在所期望的配合物。
[0216]
31
p-nmr(122mhz,cd2cl2)δ=89.41,83.71。
19
f-nmr(282mhz,cd2cl2)δ=-62.86。hr-ms(esi)m/z c
35h46
no4pir[m]+计算值768.2788,实测值768.2816。
[0217]
va-8
[0218]
根据上述标准方法,使用2-(叔丁基氯膦基)吡啶(0.25mmol)进行反应。配合物可以红色固体形式分离(196mg)。在
31
p nmr中显示出在107.59和102.97ppm(1:1.4)处的2个峰。hrms显示存在所期望的配合物。
31
p-nmr(122mhz,cd2cl2)δ=107.59,102.97。
19
f-nmr(282mhz,cd2cl2)δ=-62.83。hr-ms(esi)m/z c
34h43
n2opir[m]+计算值719.2737,实测值719.2757。
[0219]
va-9
[0220]
将相应的醇前体的thf溶液(0.25mmol,于5.0ml thf中)冷却至-78℃,并将n-buli(0.1ml 2.5m n-buli的己烷溶液;0.25mmol;1当量)滴加到连续搅拌的溶液中。加入完成
后,使溶液温热至室温,并在该温度下再搅拌30分钟。将溶液再次冷却至-78℃,并将cy2pcl(0.25mmol,1当量)加入到连续搅拌的溶液中。使混合物温热至室温,随后加热至50℃,并在该温度下保持过夜。在反应冷却至室温后,除去thf并真空干燥,将[ir(cod)cl]2(0.125mmol)和dcm(5.0ml)加入到试管中,在50℃下搅拌2小时。然后将li{al[oc(cf3)3]4}(0.275mmol)添加到反应混合物中,并在室温下搅拌过夜。将反应溶液旋转蒸发到二氧化硅上,负载于用dcm制备的二氧化硅柱上,用正庚烷/dcm:1/1层析,得到橙色固体(122mg,28%)。
[0221]1h-nmr(300mhz,cd2cl2)δ=7.40-7.12(m,4h),5.67-5.63(m,1h),5.50-5.42(m,1h),4.43-4.36(m,1h),3.39-3.30(m,1h),3.25-2.90(m,3h),2.67(s,3h),2.60-2.42(m,4h),2.42-2.18(m,2h),2.13-1.99(m,5h),1.96-1.71(m,9h),1.65-1.52(m,5h),1.45-1.01(m,12h),0.69-0.53(m,1h)。
31
p-nmr(121mhz,cd2cl2)δ=118.81。
19
f-nmr(282mhz,cd2cl2)δ=-75.74。hr-ms(esi)m/z c
37h52
nopir[m]
+
计算值750.3416,实测值750.3420。
[0222]
va-10
[0223]
根据上述方法,使用287mg[ir(cod)2]barf(0.225mmol)进行反应。配合物可以橙色固体形式分离(148mg;40%,基于[ir(cod)2]barf)。
[0224]1h-nmr(300mhz,cd2cl2):δ(ppm)=7.91-7.46(m,12h),7.21(s,1h),7.09(s,1h),6.94(s,1h),5.67-5.63(m,1h),5.46-5.41(m,1h),4.38-4.36(m,1h),3.36-3.32(m,1h),3.19-2.85(m,3h),2.64(s,3h),2.53-2.46(m,2h),2.41(s,3h),2.35(s,3h),2.31-2.18(m,2h),2.19-1.83(m,14h),1.68-1.54(m,6h),1.38-1.20(m,5h),1.14-0.97(m,5h),0.68-0.56(m,1h)。
31
p-nmr(122mhz,cd2cl2)δ=118.64。
19
f-nmr(282mhz,cd2cl2)δ=-62.87。hr-ms(esi)m/z c
38h54
nopir[m]
+
计算值764.3572,实测值764.3577。
[0225]
va-11
[0226]
根据上述方法进行反应。配合物可以橙色固体形式分离(274mg;73%,基于[ir(cod)2]barf)。
[0227]1h-nmr(300mhz,cd2cl2):δ(ppm)=7.79-7.66(m,8h),7.56(s,4h),7.29(s,1h),7.23(s,1h),7.13(s,1h),5.65(td,j=5.9,2.2hz,1h),5.46-5.40(m,1h),4.42-4.36(m,1h),3.38-3.30(m,1h),3.19-2.86(m,3h),2.65(s,3h),2.59-2.44(m,2h),2.42(s,3h),2.38-1.54(m,20h),1.46-0.98(m,21h),0.70-0.58(m,1h)。
31
p-nmr(122mhz,cd2cl2)δ(ppm)=118.67。
19
f-nmr(282mhz,cd2cl2)δ(ppm)=-62.86。hr-ms(esi)m/z c
41h60
nopir[m]
+
计算值806.4042,实测值806.4053。
[0228]
va-12
[0229]
根据上述方法,使用287mg[ir(cod)2]barf(0.225mmol)进行反应。配合物可以橙色固体形式分离(298mg;82%,基于[ir(cod)2]barf)。
[0230]1h-nmr(300mhz,cd2cl2):δ(ppm)=7.80-7.52(m,12h),7.42-7.19(m,3h),7.12(d,j=7.5hz,1h),5.65(td,j=5.6,2.6hz,1h),5.48-5.42(m,1h),4.43-4.37(m,1h),3.38-3.30(m,1h),3.21-2.89(m,3h),2.67(s,3h),2.58-2.45(m,2h),2.42(s,3h),2.38-2.16(m,2h),2.13-2.05(m,3h),2.02-1.89(m,4h),1.84(s,3h),1.81-1.72(m,2h),1.64-1.49(m,3h),1.39-1.19(m,8h),1.12-0.99(m,4h),0.68-0.56(m,1h)。
31
p-nmr(122mhz,cd2cl2)δ=118.80。
19
f-nmr(282mhz,cd2cl2)δ=-62.88。hr-ms(esi)m/z c
37h52
nopir[m]
+
计算值
750.3416,实测值750.3420。
实施例
[0231]
在金属高压釜中进行反应。反应混合物无需后处理即可通过hplc(chiralpak ic柱,95/5庚烷/乙醇,1ml/min)或sfc(oz-h柱,2.5%meoh于超临界co2,3ml/min)色谱法进行分析。
[0232]
实施例1-12:
[0233]
将ir-配合物(给定催化剂负载量)和0.64g 1-(2,2,4-三甲基-1-喹啉基)乙酮(3mmol)放入装有ptfe涂层搅拌子的8-ml高压釜样品瓶中。使用带有隔膜的螺旋盖封闭高压釜样品瓶,并用氩气冲洗(10分钟)。将六氟异丙醇(hfip,4ml)通过隔膜加入到样品瓶中。将样品瓶置于含有氩气的高压釜中,并用氩气冲洗高压釜(10分钟)。将高压釜用氢气加压(10巴),随后减压至大气压3次。此后,将高压釜加压至60巴氢气压力,并置于合适的氧化铝块中。加热至85℃后,将反应在该温度下保持给定时间。冷却至室温并减压后,将样品瓶从高压釜中取出,并通过gc-fid分析(用etoh稀释)测定反应结果,以及通过hplc分析测定对映体过量。给出典型值。
[0234]
表2:
[0235][0236]
实施例13-18:
[0237]
将ir-配合物(给定催化剂负载量)和2.56g 1-(2,2,4-三甲基-1-喹啉基)乙酮(12mmol)置于25-ml高压釜中。将高压釜用氩气冲洗(10分钟)。将六氟异丙醇(hfip,16ml)
加入至高压釜中。将高压釜用氢气加压(10巴),随后减压至大气压3次。此后,将高压釜加压至60巴氢气压力,并置于合适的氧化铝块中。加热至85℃后,将反应在该温度下保持给定时间。冷却至室温并减压后,通过gc-fid分析(用etoh稀释)测定反应结果,并通过hplc分析测定对映体过量。
[0238]
表3:
[0239][0240]
实施例19-48
[0241]
将ir-配合物va-1(给定的催化剂负载量)和0.64g 1-(2,2,4-三甲基-1-喹啉基)乙酮(3mmol,用庚烷:水洗涤+结晶进行纯化)放置于8ml装有ptfe涂层搅拌子的高压釜样品瓶中。使用带有隔膜的螺旋盖封闭高压釜样品瓶,并用氩气冲洗(10分钟)。将六氟异丙醇(hfip,4ml)和添加剂(给定的负载量)通过隔膜添加至样品瓶中。将样品瓶放置于含有氩气的高压釜中,并用氩气冲洗高压釜(10分钟)。将高压釜用氢气(10巴)加压,随后减压至大气压3次。此后,将高压釜加压至60巴氢气压力,并置入合适的氧化铝块中。加热至85℃后,将反应在此温度下保持给定的时间。冷却至室温并减压后,将样品瓶从高压釜中取出,并通过gc-fid分析(用etoh稀释)测定反应结果,以及通过hplc分析测定对映体过量。给出了典型值。
[0242]
表4:
[0243]
[0244][0245]
实施例49-54:
[0246]
将ir-配合物va-1(给定的催化剂负载量)和1-(2,2,4-三甲基-1-喹啉基)乙酮(给定量;用庚烷:水洗涤+结晶进行纯化)放置于25ml高压釜。用氩气冲洗高压釜(10分钟)。将六氟异丙醇(1.33ml/mmol 1-(2,2,4-三甲基-1-喹啉基)乙酮)和添加剂(给定负载量)添加至高压釜。将高压釜用氢气(10巴)加压,随后减压至大气压3次。此后,将高压釜加压至60巴氢气压力,并置入合适的氧化铝块中。加热至85℃后,将反应在此温度下保持给定时间。冷却至室温并减压后,通过gc-fid分析(用etoh稀释)测定反应结果,并通过hplc分析测定对映体过量。
[0247]
表5:
[0248][0249][0250]
实施例55-56:
[0251]
将ir配合物(给定标识符和催化剂负载量)和0.64g 1-(2,2,4-三甲基-1-喹啉基)乙酮(3mmol,用庚烷:水洗涤+结晶进行纯化)放置于8ml装有ptfe涂层搅拌子的高压釜样品瓶中。使用带有隔膜的螺旋盖封闭高压釜样品瓶,并用氩气冲洗(10分钟)。将六氟异丙醇(hfip,4ml)和bf3*oet2(1mol%相对于1-(2,2,4-三甲基-1-喹啉基)乙酮)通过隔膜添加至样品瓶中。将样品瓶放置于含有氩气的高压釜中,并用氩气冲洗高压釜(10分钟)。将高压釜用氢气(10巴)加压,随后减压至大气压3次。此后,将高压釜加压至60巴氢气压力,并置入合适的氧化铝块中。加热至85℃后,将反应在此温度下保持给定时间。冷却至室温并减压后,将样品瓶从高压釜中取出,并通过gc-fid分析(用etoh稀释)测定反应结果,以及通过hplc分析测定对映体过量。给出典型值。
[0252]
表6:
[0253][0254]
实施例57-60:
[0255]
将ir-配合物va-1(0.02mol%,0.6μmol)和0.64g 1-(2,2,4-三甲基-1-喹啉基)乙酮(3mmol,用庚烷:水洗涤+结晶进行纯化)放置于8ml装有ptfe涂层搅拌子的高压釜样品瓶中。使用带有隔膜的螺旋盖封闭高压釜样品瓶,并用氩气冲洗(10分钟)。将2,2,2-三氟乙醇(tfe,4ml)和bf3*oet2(给定负载量)通过隔膜添加至样品瓶中。将样品瓶放置于含有氩气的高压釜中,并用氩气冲洗高压釜(10分钟)。将高压釜用氢气(10巴)加压,随后减压至大气压3次。此后,将高压釜加压至60巴氢气压力,并置入合适的氧化铝块中。加热至85℃后,将反应在此温度下保持3小时。冷却至室温并减压后,将样品瓶从高压釜中取出,并通过gc-fid分析(用etoh稀释)测定反应结果,以及通过hplc分析测定对映体过量。给出典型值。
[0256]
表7:
[0257]
实施例bf3*oet2(mol%)转化率gc(%a/a)57-《1581865938860582
[0258]
比较例
[0259]
将ir-配合物(给定催化剂负载量)和0.64g 1-(2,2,4-三甲基-1-喹啉基)乙酮(3mmol)置入装有ptfe涂层搅拌子的8-ml高压釜样品瓶中。使用带有隔膜的螺旋盖封闭高压釜样品瓶,并用氩气冲洗(10分钟)。将六氟异丙醇(hfip,4ml)通过隔膜加入到样品瓶中。将样品瓶置于含有氩气的高压釜中,并用氩气冲洗高压釜(10分钟)。将高压釜用氢气加压(10巴),并随后减压至大气压3次。此后,将高压釜加压至60巴氢气压力,并置于合适的氧化铝块中。加热至85℃后,将反应在该温度下保持给定时间。冷却至室温并减压后,将样品瓶从高压釜中取出,并通过gc-fid分析(用etoh稀释)测定反应结果,以及通过hplc分析测定对映体过量。
[0260]
表8:
[0261]
实施例催化剂催化剂负载量(mol%)时间(h)转化率17va-10.01650.818va-20.01653.415va-10.011793.113va-100.011782.561va-100.02516.59862va-130.11699.263ir催化剂(1)0.11684
[0262]
这组实验结果显示了配合物va-1和va-2的优越性。使用0.01mol%的催化剂6小时后,va-2的转化率略高于va-1。在使用0.01mol%的催化剂17小时后,va-1的转化率明显高于va-10(=wo2019/185541 a1的va-10)。尽管仅使用了1/4的催化剂量,但va-10的性能与va-13(=wo2019/185541a1的va-1)相似。因此必须认为其优于va-13。与de112015001290t5的ir催化剂(i)相比,va-13的转化率更高。此外,与de 112015001290 t5的ir催化剂(1)(81.7%ee)相比,所有催化剂va都具有更优异的对映选择性(》95%ee)。相比于de 112015001290 t5的ir催化剂(1),催化剂va-1即使在仅十分之一的催化剂负载量(0.01相比于0.1mol%)下,也具有更高的转化率(93.1相比于84%)。
[0263]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1