一种2,3-吡啶二羧酸酯衍生物中间体及2,3-吡啶二羧酸酯衍生物的制备方法与流程
1.本发明涉及农药化学领域,尤其涉及一种2,3-吡啶二羧酸酯衍生物中间体及2,3-吡啶二羧酸酯衍生物的制备方法。
背景技术:
2.咪唑啉酮类化合物作为高效性除草剂,作用机制主要抑制乙酸羟酸合成酶(ahas)的活性,影响3种支链氨基酸——缬氨酸、亮氨酸与异亮氨酸的生物合成,最终破坏蛋白质的合成,干扰dna合成及细胞分裂和生长。而甲氧咪草烟,甲基咪草烟,乙基咪草烟是咪唑啉酮类除草剂的重要组成部分。
3.专利us4948896a和us5047542a中公开了马来酸二烷基酯衍生物合成吡啶-2,3-二羧酸酯的方法,总体收率66-88%,产品纯度69-93%,收率和纯度均不够理想,此外还存在有机三废高的问题,难以解决。
4.中国发明专利cn1062724a、cn1057456a分别公开了由氯代马来酸酯、二氯代马来酸二烷基酯制备吡啶-2,3-二羧酸二烷基酯的方法,其原料简单易得,但是转化率低,而且氯代马来酸酯遇到高热和碱性物质会发生分解,中间体稳定性较差,反应时间长达16~72小时,一定程度上增加工业化的难度。
5.美国专利us4816588a和us5597924a公开了用喹啉衍生物经过过氧化物氧化得到产品,氧化本身属于危险工艺,具有较高的危险性,而且收率很低仅为48~69%,原子利用率差。
6.美国专利us5334576a和us4798619a公开了以苯胺类衍生物为原料制备吡啶-2,3-二羧酸酯的方法,收率较低且危险。
7.中国发明专利cn104447527a公开了一种吡啶-2,3-二羧酸二烷基酯的方法,收率虽然能做到82~96%,但是需要用到危险的催化剂氨基钠、钠氢或三苯甲基钠,不利于工业化生产,另外反应温度高,引起副反应多,并且不易控制反应,工艺复杂。
8.因此,作为咪唑啉酮类化合物合成的重要中间体,现有的吡啶-2,3-二羧酸酯合成方法均不够理想,亟需一种反应步骤短、原料易得、反应温和、产品收率和纯度较高的吡啶-2,3-二羧酸酯合成方法。
技术实现要素:
9.本发明要解决的技术问题是:提供一种反应步骤短、原料易得、反应温和、产品收率和纯度较高的吡啶-2,3-二羧酸酯中间体及其合成方法。
10.本发明解决上述技术问题的技术方案如下:
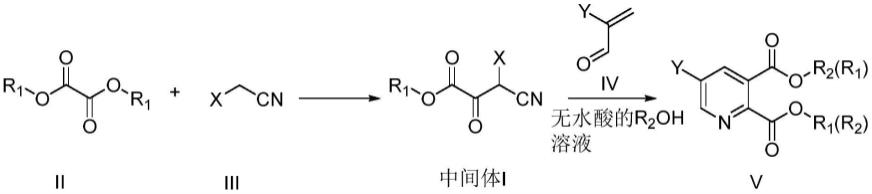
11.一种吡啶-2,3-二羧酸酯衍生物的中间体,其结构如式ⅰ所示:
[0012][0013]
其中,r1选自c
1~6
烷基;x选自卤素,c
1~4
的烷氧基或c
1~4
的烷硫基。
[0014]
本发明的第二目的是提供一种基于上述中间体的吡啶-2,3-二羧酸酯衍生物的合成方法,包括如下步骤:
[0015]
(1)将通式ⅱ化合物和通式ⅲ化合物溶于有机溶剂中,加入强碱反应,反应结束后用无水酸的醇溶液酸化处理得到含有中间体i的酸化反应液;将酸化反应液直接投入下步反应或分离得到中间体i后投入下步反应;
[0016]
(2)将含有中间体i的酸化反应液热处理,然后加入通式iv化合物;或将中间体i加入无水酸的醇溶液中,再加入通式iv化合物;反应结束后,经后处理即得到通式
ⅴ
;
[0017]
具体反应式如下:
[0018][0019]
其中,r1和r2独立的选自c
1~6
烷基;x选自卤素,c
1~4
的烷氧基或c
1~4
的烷硫基;
[0020]
y选自氢、卤素、c
1~6
烷基,c
1~4
烷氧羰基,氨基羰基,苯硫基,苯氧基或苯基;以及被一个或多个卤素、羟基、c
1~4
的烷氧基或c
1~4
的烷硫基取代的c
1~6
烷基,c
1~4
烷氧羰基,氨基羰基,苯硫基,苯氧基或苯基;当r1与r2不相同时,得到的产物为混合物。
[0021]
优选的,所述步骤(1)中的有机溶剂为醇类,如甲醇,乙醇或正丁醇;芳烃类,如甲苯或二甲苯等。
[0022]
优选的,所述步骤(1)中的反应温度为-10~80℃,进一步的5~50℃。
[0023]
优选的,所述步骤(1)中的强碱选自氢氧化钠、氢氧化钾、氢氧化锂、碳酸钠、碳酸钾、碳酸锂、甲醇钠、甲醇钾、甲醇锂、乙醇钠、乙醇钾、乙醇锂、异丙醇钠、异丙醇钾、异丙醇、锂叔丁醇钠、叔丁醇钾、叔丁醇锂、氢化钠、氨基钠、三乙胺或吡啶等各种常见无机碱和三级有机碱。
[0024]
优选的,所述步骤(1)中的无水酸的醇溶液为盐酸甲醇溶液、盐酸乙醇溶液、硫酸甲醇溶液或硫酸乙醇溶液等。
[0025]
优选的,所述步骤(1)中的通式ⅱ、通式ⅲ、强碱和无水酸的摩尔比为1:1.0~5.0:1.0~5.0:1.0~15.0;
[0026]
进一步的,述步骤(1)中的通式ⅱ、通式ⅲ、强碱和无水酸的摩尔比为1:1.0~1.5:1.0~1.5:1.0~4.0。
[0027]
更进一步的,述步骤(1)中的通式ⅱ、通式ⅲ、强碱和无水酸的摩尔比为1:1.01~1.3:1.0~1.5:1.3~2.5。
[0028]
优选的,所述步骤(2)中反应温度为-10~120℃;进一步的40~90℃。
[0029]
优选的,所述步骤(2)中的中间体i、通式iv、酸的投料摩尔比为1:1.0~2.0:0~2。
[0030]
进一步的,所述步骤(2)中的中间体i、通式iv、酸的投料摩尔比为1:1.1~1.2:0~1.2。
[0031]
本发明中化合物的中文命名与结构式有冲突的,以结构式为准;结构式有明显错误的除外。
[0032]
本发明提供的2,3-吡啶二羧酸酯衍生物中间体及2,3-吡啶二羧酸酯衍生物的制备方法,两步反应可以通过一锅煮的方法进行,第二步生成游离的氨提供吡啶成环的氨源,整个工艺安全环保,原子利用率高,经济高效,适合工业化生产。
具体实施方式
[0033]
以下结合实例说明本发明,但不限制本发明。在本领域内,技术人员对本发明所做的简单替换或改进均属于本发明所保护的技术方案内。
[0034]
实施例1:
[0035]
①
3-氯-3-氰基-2-氧代丙酸甲酯的制备:
[0036][0037]
将118.09g(1mol)的草酸二甲酯和180.1g的30%甲醇钠甲醇溶液(1mol)室温搅拌溶解,控制15~20℃缓慢滴加90.60g的氯乙腈(1.2mol),滴加用时3h,滴加完毕后保温搅拌1h。取样合格后降温至5~10℃,缓慢加入182.5g的30%盐酸甲醇溶液(1.5mol),酸化到ph值小于4,过滤,蒸干甲醇得到160g的3-氯-3-氰基-2-氧代丙酸甲酯,定性含量94%,收率93.4%。1h nmr(400mhz,dmso)δ7.07(s,1h),3.83(s,3h).
[0038]
②
5-甲基吡啶-2,3-二羧酸甲酯的制备:
[0039][0040]
将步骤
①
得到的160g的94%的3-氯-3-氰基-2-氧代丙酸甲酯(0.93mol)加入136g的30%盐酸甲醇溶液(1.1mol),升温至65℃,缓慢滴加78.2g的2-甲基丙烯醛(1.1mol),用时5h,滴加完毕后保温搅拌5h。取样合格后蒸干甲醇,加水溶液,二氯乙烷萃取,蒸干溶剂得到黄色固体185.03g,定性含量95%,收率90.3%,1h nmr(400mhz,dmso)δ8.67-8.62(m,1h),8.11(dd,j=1.9,0.7hz,1h),3.85(s,6h),2.41(s,3h)。
[0041]
实施例2:
[0042]
①
3-氯-3-氰基-2-氧代丙酸甲酯的制备:
[0043][0044]
将118.09g的草酸二甲酯(1mol)和270.1g的30%甲醇钠甲醇溶液(1.5mol)室温搅拌溶解,控制15~20℃缓慢滴加98.15g的氯乙腈(1.3mol),滴加用时3h,滴加完毕后保温搅
拌1h。取样合格后降温至5-10℃,缓慢加入304.17g的30%盐酸甲醇溶液(2.5mol),酸化到ph值小于4,得到593.89g的3-氯-3-氰基-2-氧代丙酸甲酯的酸化反应液,不分离直接进行下一步反应。
[0045]
②
5-甲基吡啶-2,3-二羧酸甲酯的制备:
[0046][0047]
将实施例2步骤
①
得到的593.89g的3-氯-3-氰基-2-氧代丙酸甲酯的酸化反应液升温至60℃,缓慢滴加78.2g的2-甲基丙烯醛(1.1mol),用时5h,滴加完毕后保温搅拌5h。取样合格后蒸干甲醇,加水溶液,二氯乙烷萃取,蒸干溶剂得到黄色固体194.56g,定性含量96%,两步总收率89.3%。1h nmr(400mhz,dmso)δ8.67-8.62(m,1h),8.11(dd,j=1.9,0.7hz,1h),3.85(s,6h),2.41(s,3h).
[0048]
实施例3:
[0049]
①
3-甲氧基-3-氰基-2-氧代丙酸乙酯的制备:
[0050][0051]
将146.14g的草酸二乙酯(1mol)和102.1g的乙醇钠(1.5mol)溶于500g甲苯中,控制30~35℃缓慢滴加78.1g的氯乙腈(1.03mol),滴加用时2h,滴加完毕后保温搅拌2h。取样合格后降温至5-10℃,缓慢加入158.28g的30%盐酸乙醇溶液(1.3mol),酸化到ph值小于4,过滤,蒸干溶剂得到154.04g的3-甲氧基-3-氰基-2-氧代丙酸乙酯,定性含量95%,收率85.5%。1h nmr(400mhz,dmso)δ10.85(s,1h),4.28-4.22(m,2h),3.71(s,3h),1.26(t,j=7.1hz,3h).
[0052]
②
5-乙基吡啶-2,3-二羧酸乙酯的制备:
[0053][0054]
将实施例3步骤
①
得到的154.04g的95%的3-甲氧基-3-氰基-2-氧代丙酸乙酯加入117.5g的30%盐酸乙醇溶液(0.966mol),升温至80℃,缓慢滴加79.11g的2-乙基丙烯醛(0.94mol),用时3h,滴加完毕后保温搅拌6h。取样合格后蒸干甲醇,加水溶液,二氯甲烷萃取,蒸干溶剂得到黄色固体197.66g,定性含量94%,收率86.5%。1h nmr(400mhz,dmso)δ8.69(d,j=2.0hz,1h),8.13(d,j=2.0hz,1h),4.43-4.23(m,4h),2.76(q,j=7.6hz,2h),1.33(td,j=7.1,1.5hz,6h),1.27-1.17(m,3h).
[0055]
以上所述的仅是本发明的优选实施方式,应当指出,对于本领域的普通技术人员来说,在不脱离本发明创造构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1