一种通过Meerwein芳基化反应合成2-氯-4-氟苯甲酸的方法与流程
一种通过meerwein芳基化反应合成2-氯-4-氟苯甲酸的方法
技术领域
1.本发明涉及有机合成领域,尤其涉及一种通过meerwein芳基化反应合成2-氯-4-氟苯甲酸的方法。
背景技术:
2.2-氯-4-氟苯甲酸是一种重要的精细化工中间体,它广泛应用在医药和农药领域,特别地,它是除草剂苯嘧磺草胺的关键中间体,该除草剂具有巨大的市场潜力。
3.关于2-氯-4-氟苯甲酸的合成技术,参考国内外文献主要有以下几种方法:
4.1、目前国内的传统合成工艺,通常采用重铬酸盐为氧化剂,将2-氯-4-氟甲苯氧化为2-氯-4-氟苯甲酸,但是重铬酸盐毒性大,对环境污染严重,且产品收率较低。
[0005][0006]
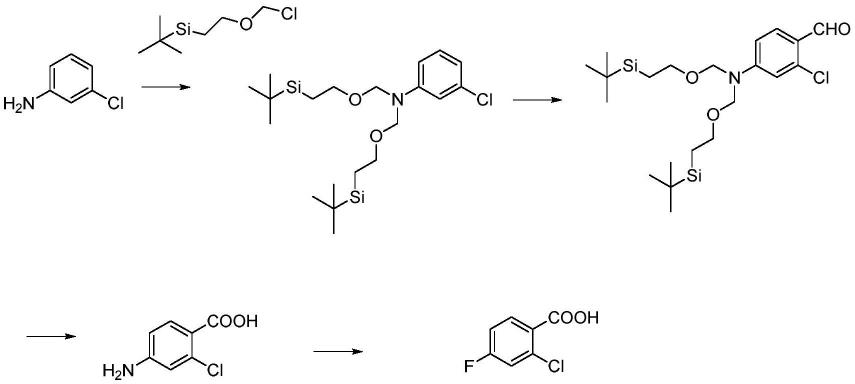
2、中国专利cn105732357a以间氯苯胺为原料,先用2-(三甲硅烷基)乙氧甲基氯将氨基保护,其次通过vilsmeier-haack反应实现甲酰化,然后加入双氧水和三苯基膦氯化铑反应生成2-氯-4-氨基苯甲酸,最后加入双氧水和氟化钾及磷钨酸杂多酸铵盐反应得到2-氯-4-氟苯甲酸。该方法虽然原料简单易得,但用到保护/脱保护的策略,同时氧化步骤还用到贵金属铑催化剂,增加了成本。
[0007][0008]
3、中国cn107556289a以2-氯-4-氟苯甲醛为原料,加入亚氯酸钠氧化为2-氯-4-氟苯甲酸。该方法原料昂贵,且亚氯酸钠为强氧化剂,具有爆炸风险。
[0009][0010]
4、mallia等人在beilstein journal of organic chemistry 2016,12,1503-1511中以2-氯-4-氟碘苯为原料,采用醋酸钯催化,与一氧化碳进行插羰反应得到2-氯-4-氟苯甲酸。该方法用到昂贵的金属催化剂,成本较高,且一氧化碳具有泄露风险,危险性大。
[0011][0012]
综上所述,现有的2-氯-4-氟苯甲酸制备方法分别存在着原料昂贵、反应试剂危险性大、催化剂价格昂贵、合成产物收率低等问题。
技术实现要素:
[0013]
本发明要解决的技术问题是:提供一种反应温和、步骤少、收率高,且反应过程不使用剧毒试剂和昂贵催化剂的合成2-氯-4-氟苯甲酸的方法。
[0014]
本发明解决上述技术问题的技术方案如下:
[0015]
一种通过meerwein芳基化反应合成2-氯-4-氟苯甲酸的方法,包括如下步骤:
[0016]
(1)2-氯-4-氟苯胺在无水氯化铜的催化下与亚硝酸叔丁酯、1,1-二氯乙烯发生meerwein芳基化反应生成2-氯-4-氟三氯甲苯;
[0017]
(2)2-氯-4-氟三氯甲苯在酸性条件下水解生成2-氯-4-氟苯甲酸;具体反应式如下:
[0018][0019]
优选的,所述步骤(1)中2-氯-4-氟苯胺、1,1-二氯乙烯、亚硝酸叔丁酯与无水氯化铜的摩尔比为1:0.5~30.0:0.5~5.0:0.5~2.0。
[0020]
进一步的,所述步骤(1)中2-氯-4-氟苯胺、1,1-二氯乙烯、亚硝酸叔丁酯与无水氯化铜的摩尔比为1:1.0~20.0:1.0~3.0:1.0~2.0。
[0021]
更进一步的,所述步骤(1)中2-氯-4-氟苯胺、1,1-二氯乙烯、亚硝酸叔丁酯与无水氯化铜的摩尔比为1:10.0~15.0:1.2~1.5:1.1~1.3。
[0022]
优选的,所述步骤(1)中反应温度为-10~20℃。
[0023]
进一步的,所述步骤(1)中反应温度为-5~10℃。
[0024]
优选的,所述步骤(2)中所用的酸包括盐酸、硫酸、磷酸、醋酸等常用无机酸或有机酸。
[0025]
优选的,所述步骤(2)中2-氯-4-氟三氯甲苯与酸的摩尔比为1:0.5~20.0。
[0026]
进一步的,所述步骤(2)中2-氯-4-氟三氯甲苯与酸的摩尔比为1:2.0~10.0。
[0027]
更进一步的,所述步骤(2)中2-氯-4-氟三氯甲苯与酸的摩尔比为1:5.0~10.0。
[0028]
优选的,所述步骤(2)反应温度为60~100℃;进一步的,所述步骤(2)反应温度为70~80℃。
[0029]
本发明中化合物的中文命名与结构式有冲突的,以结构式为准;结构式有明显错误的除外。
[0030]
本发明提供的通过meerwein芳基化反应合成2-氯-4-氟苯甲酸的方法优势在于避免了昂贵的贵金属催化剂和剧毒试剂的使用,所用试剂对环境友好,降低了成本,简化了工艺,收率较高,克服了现有技术的不足,适宜大规模工业化生产。
具体实施方式
[0031]
以下结合实例说明本发明,但不限制本发明。在本领域内,技术人员对本发明所做的简单替换或改进均属于本发明所保护的技术方案内。
[0032]
实施例1:
[0033][0034]
在2000ml四口瓶中,依次加入100g乙腈,43.23g无水氯化铜(分子量134.45,321.52mmol,1.3eq),359.63g 1,1-二氯乙烯(分子量96.94,3709.81mmol,15eq)和38.26g亚硝酸叔丁酯(分子量103.12,370.98mmol,1.5eq),反应体系降温至5℃,缓慢滴加108g2-氯-4氟苯胺的乙腈溶液(分子量145.56,247.32mmol,1eq,36g 2-氯-4-氟苯胺溶于72g乙腈),约1h加完。加完后,将反应液移至室温搅拌16h;抽滤反应液,滤除铜盐。滤液负压脱除乙腈和1,1-二氯乙烯,然后加入100g乙酸乙酯稀释,乙酸乙酯相依次用80g 10%盐酸洗涤,60g水洗涤。有机相负压蒸干得粗品,粗品用40g正己烷打浆得到49.80g类白色固体,液相色谱检测纯度91.18%,即为2-氯-4-氟三氯甲苯(分子量247.90,理论得到61.31g),质量收率81.22%。
[0035]
实施例2:
[0036]
在2000ml四口瓶中,依次加入100g乙腈,36.58g无水氯化铜(分子量134.45,272.05mmol,1.1eq),239.75g 1,1-二氯乙烯(分子量96.94,2473.2mmol,10eq)和30.60g亚硝酸叔丁酯(分子量103.12,296.78mmol,1.2eq),反应体系降温至0℃,缓慢滴加108g 2-氯-4氟苯胺的乙腈溶液(分子量145.56,247.32mmol,1eq,36g 2-氯-4-氟苯胺溶于72g乙腈),约1h加完。加完后,将反应液移至室温搅拌12h;抽滤反应液,滤除铜盐。滤液负压脱除乙腈和1,1-二氯乙烯,然后加入100g乙酸乙酯稀释,乙酸乙酯相依次用80g 10%盐酸洗涤,60g水洗涤。有机相负压蒸干得粗品,粗品用40g正己烷打浆得到52.21g类白色固体,液相色谱检测纯度90.15%,即为2-氯-4-氟三氯甲苯(分子量247.90,理论得到61.31g),质量收率85.16%。
[0037]
实施例3:
[0038][0039]
在500ml四口瓶中依次加入96.81g 98%浓硫酸(分子量98,968.13mmol,5eq)和48g 2-氯-4-氟三氯甲苯(分子量247.90,193.63mmol,1.0eq),将反应混合物升温至80℃搅拌7h。将反应液冷至室温,然后缓慢地倒入300g冰水中,倒完后有大量固体析出并搅拌0.5h,过滤,滤饼用100g水淋洗,挖出滤饼并放置70℃烘箱中烘干,称重得到30.47g白色固体,液相色谱检测纯度94.15%,即为2-氯-4氟苯甲酸(分子量174.56,理论得到33.80g),质量收率90.15%。
[0040]
实施例4:
[0041]
在500ml四口瓶中依次加入193.62g 98%浓硫酸(分子量98,1936.3mmol,10eq)和48g2-氯-4-氟三氯甲苯(分子量247.90,193.63mmol,1.0eq),将反应混合物升温至90℃搅拌7h。将反应液冷至室温,然后缓慢地倒入500g冰水中,倒完后有大量固体析出并搅拌0.5h,过滤,滤饼用100g水淋洗,挖出滤饼并放置60℃烘箱中烘干,称重得到30.66g白色固体,液相色谱检测纯度93.78%,即为2-氯-4氟苯甲酸(分子量174.56,理论得到33.80g),质量收率90.71%。
[0042]
(1h-nmr(dmso-d6)δ:13.45(s,br,1h),7.91(dd,1h),7.56(dd,1h),7.32(ddd,1h))
[0043]
以上所述的仅是本发明的优选实施方式,应当指出,对于本领域的普通技术人员来说,在不脱离本发明创造构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1