一种去氢中美菊素C衍生物、其制备方法及应用与流程
一种去氢中美菊素c衍生物、其制备方法及应用
技术领域
1.本发明涉及一种去氢中美菊素c衍生物、其制备方法及应用。
背景技术:
2.核转录因子κb(nuclear factor kappa-b,nf-κb)最先由baltimore和sen发现,是一种促炎转录因子,它在细胞的发育、分化和各种病理状态中起着重要的作用。越来越多的研究表明nf-κb的异常激活在炎症和癌症中起着重要的作用。nf-κb能调节许多炎症因子的表达,例如环氧合酶-2(cox-2)、趋化因子、黏附分子、白细胞介素-1、白细胞介素-6、5-脂氧化酶(5-lox)、基质金属蛋白酶家族(mmps)、肿瘤坏死因子(tnf)和血管内皮生长因子(vegf)等等与肿瘤生长相关的因子。这也为nf-κb参与的炎症在癌症中起到的作用提供了分子生物学基础。在哺乳动物中nf-κb主要由五个亚基组成:nf-κb1(p105/p50)、nf-κb2(p100/p52)、c-rel、rela(p65)和relb。这些亚基形成的同源二聚体和异源二聚体共同调控位于nf-κb信号通路下游靶基因的表达。在正常情况下,nf-κb会与调控其激活的内源性抑制蛋白iκb(通常为iκbα,iκbβ,iκbε,iκbns)结合存在于细胞质当中。
3.尽管目前已有超过15条nf-κb信号通路激活途径已被报道,但主要可以归为经典的激活途径和非经典的激活途径两种。经典途径由p105/p50的激活而启动,非经典激活途径由p100/p52的激活而启动。在经典途径中负责调控的是ikkβ亚基及内源性抑制蛋白家族iκbs,而非经典途径则由ikkα二聚体和nik激酶负责调控。在经典激活途径中,ikkβ通过磷酸化iκbα的两个保守的n-端丝氨酸残基(ser32/36)启动激活途径。iκbα的磷酸化会触发e2、e3连接酶的多聚泛素化导致iκbα被26s蛋白酶体降解。iκbα被降解后会释放nf-κb二聚体(p65/p50)进入细胞核从而激活nf-κb下游靶基因的表达。nf-κb的p65亚基在经历一系列的翻译修饰(包括被ikk磷酸化的ser536位点)后才能激活nf-κb信号通路。在非经典途径的激活中,通常涉及到的是relb和由p100前体蛋白处理后形成的p52两者结合后形成的p52-relb异源二聚体,随后异源二聚体入核激活下游靶基因的表达。由于ikk在nf-κb信号通路中起到的关键作用,ikk已经成为治疗癌症的靶标之一。
技术实现要素:
4.本发明所要解决的技术问题是寻找一种抑制mda-mb-231和bt-549肿瘤细胞株的生长的化合物,为此,本发明提供了一种去氢中美菊素c衍生物、其制备方法及应用,既可以阻断nf-κb信号通路的激活,又可以将其应用在制备治疗三阴性乳腺癌药物中,抑制mda-mb-231和bt-549肿瘤细胞株的生长。
5.本发明主要是通过以下技术方案解决上述技术问题的。
6.本发明提供了一种式t所示化合物、其药学上可接受的盐、其溶剂合物或其药学上可接受的盐的溶剂合物,
[0007][0008]
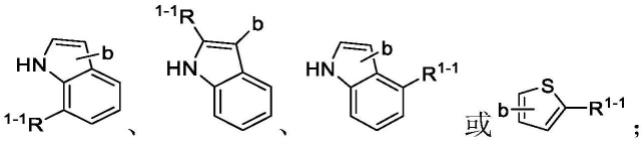
其中,r1为5~10元杂芳基或被一个或两个r
1-1
取代的5~10元杂芳基;
[0009]r1-1
独立地为卤素、羟基、羧基、甲磺酰基、硝基、氰基、c1~c4烷基、被r
1-1-1
取代的c1~c4烷基、c1~c4烷氧基、r
1-1-2
取代的c1~c4烷氧基、苄氧基或
[0010]r1-1-1
和r
1-1-2
独立地为卤素;
[0011]rl
独立地为c1~c4烷基;
[0012]
其中,所述的5~10元杂芳基中,杂原子选自n、s和o中的一种或多种;杂原子的个数为一个。
[0013]
在所述的制备方法的某一方案中,某些技术特征的定义如下所述,其他的技术特征的定义如上任一方案所述(以下简称“在某一方案中”):
[0014]
当r1为5~10元杂芳基时,所述的5~10元杂芳基可为吲哚基、吡咯基、噻吩基、氧茚基或呋喃基,例如吲哚基、氧茚基、吡咯基或噻吩基。
[0015]
在某一方案中,当r1为被一个或两个r
1-1
取代的5~10元杂芳基时,所述的5~10元杂芳基可为吲哚基、吡咯基、噻吩基、氧茚基或呋喃基,例如吲哚基、氧茚基、吡咯基或噻吩基。
[0016]
在某一方案中,当r
1-1
为c1~c4烷基时,所述的c1~c4烷基可为甲基、乙基,丙基、异丙基、正丁基、异丁基或叔丁基,例如甲基。
[0017]
在某一方案中,当r
1-1
为被r
1-1-1
取代的c1~c4烷基时,所述的c1~c4烷基可为甲基、乙基,丙基、异丙基、正丁基、异丁基或叔丁基,例如甲基。
[0018]
在某一方案中,所述的r
1-1-1
为f、cl、br或i,例如f。
[0019]
在某一方案中,当r
1-1
为被r
1-1-1
取代的c1~c4烷基时,所述的被r
1-1-1
取代的c1~c4烷基可为-cf3。
[0020]
在某一方案中,当r
1-1
为c1~c4烷氧基时,所述的c1~c4烷氧基可为甲氧基、乙氧基,丙氧基、异丙氧基、正丁氧基、异丁氧基或叔丁氧基,例如甲氧基。
[0021]
在某一方案中,当r
1-1
为被r
1-1-2
取代的c1~c4烷氧基时,所述的c1~c4烷氧基可为甲氧基、乙氧基,丙氧基、异丙氧基、正丁氧基、异丁氧基或叔丁氧基,例如甲氧基。
[0022]
在某一方案中,所述的r
1-1-2
为f、cl、br或i,例如f。
[0023]
在某一方案中,当r
1-1
为被r
1-1-2
取代的c1~c4烷氧基时,所述的被r
1-1-2
取代的c1~c4烷氧基可为-ocf3或-och2f。
[0024]
在某一方案中,所述的r
l
可为甲基、乙基,丙基、异丙基、正丁基、异丁基或叔丁基,例如甲基。
[0025]
在某一方案中,当r
1-1
为时,所述的为为例如
[0026]
在某一方案中,当r1为被一个或两个r
1-1
取代的5~10元杂芳基时,所述的被一个或两个r
1-1
取代的5~10元杂芳基可为取代的5~10元杂芳基可为(b为与连接的位点)。
[0027]
在某一方案中,当r1为5~10元杂芳基或被一个或两个r
1-1
取代的5~10元杂芳基时,所述的5~10元杂芳基为时,所述的5~10元杂芳基为
[0028]
在某一方案中,r1为
[0029]
在某一方案中,所述的如式t所示化合物中可为或二者的混合物。
[0030]
在某一方案中,所述的如式t所示化合物可如下任一所示:
[0031]
[0032]
本发明还提供了一种上述的如式t所示的化合物的制备方法,其包括下述步骤,有机溶剂中,催化剂作用下,将式c所示化合物与r1h进行加成反应,得到式t所示化合物即可;
[0033][0034]
其中,r1的定义如上所述。
[0035]
在所述的加成反应中,所述的催化剂可为本领域该类反应的常规催化剂,例如四氧化锆。
[0036]
在所述的加成反应中,所示的r1h可为h可为
[0037]
在所述的加成反应中,所述的有机溶剂可为本领域该类反应常规的有机溶剂,例如二氯甲烷。
[0038]
在所述的加成反应中,所述的式c所示化合物与所述的r1h的摩尔比可为1:0.5~1:2,例如1:1。
[0039]
在所述的加成反应中,所述的式c所示化合物与所述的催化剂的摩尔比为1:0.1~1:1,例如1:0.3。
[0040]
所述的反应的温度可为本领域该类反应常规的温度,优选地20~40℃,例如25℃。
[0041]
所述的反应的进程可采用本领域常规的检测方式进行监测,如薄层色谱(tlc)、气相色谱(gc)、核磁共振波谱(nmr)或高效液相色谱(hplc)等。
[0042]
所述的反应的时间以所述的反应完全为准,例如10~50min,又例如20min。
[0043]
所述的反应的后处理步骤可为本领域该类反应常规的后处理步骤,例如:减压蒸馏、过滤或柱层析,得到所述的式t所示化合物即可。
[0044]
所述的如式t所示化合物的制备方法,还可进一步包括下述步骤:有机溶剂中,将式a所示化合物与脱氢试剂进行消除反应,得到所述的式c所示化合物即可;
[0045][0046]
在所述的消除反应中,所述的有机溶剂可为本领域该类反应的常规溶剂,例如二氯甲烷。
[0047]
在所述的消除反应,所述的脱氢试剂可为本领域该类反应的常规脱氢试剂,例如戴斯-马丁过碘烷。
[0048]
在所述的消除反应中,所述的式a所示化合物与所述的脱氢试剂的摩尔比可为0.5~2,例如1。
[0049]
在所述的消除反应中,所述的式a所示化合物与所述的有机溶剂的质量体积比可为40g/l~90g/l,例如60g/l。
[0050]
所述的消除反应的温度可为本领域该类反应常规的温度,优选地20~40℃,例如25℃。
[0051]
所述的消除反应的进程可采用本领域常规的检测方式进行监测,如薄层色谱(tlc)、气相色谱(gc)、核磁共振波谱(nmr)或高效液相色谱(hplc)等。
[0052]
所述的消除反应的时间以所述的消除反应完全为准,例如0.1~4h,例如1小时。
[0053]
所述的消除反应的后处理步骤可为本领域该类反应常规的后处理步骤,例如:过
滤,柱层析,得到所述的式a所示化合物即可。
[0054]
所述的如式t所示化合物的制备方法,还可进一步包括下述步骤:有机溶剂中,催化剂的作用下,将式s所示化合物与氧化试剂进行氧化反应,得到所述的式a所示化合物即可;
[0055][0056]
在所述的氧化反应中,所述的有机溶剂可为本领域该类反应的常规溶剂,例如二氯甲烷。
[0057]
在所述的氧化反应中,所述的催化剂可为二氧化硒(seo2)。
[0058]
在所述的氧化反应中,所述的氧化试剂可为本领域该类反应的常规氧化试剂,例如过氧化叔丁醇(t-buooh)。
[0059]
在所述的氧化反应中,所述的式s所示化合物与所述的氧化试剂的摩尔比可为0.1~1,例如0.5。
[0060]
在所述的氧化反应中,所述的式s所示化合物与所述的催化剂的摩尔比可为1~4,例如2。
[0061]
在所述的氧化反应中,所述的式s所示化合物与所述的有机溶剂的质量体积比可为5g/l~20g/l,例如13g/l。
[0062]
所述的氧化反应的温度可为本领域该类反应常规的温度,优选地20~40℃,例如25℃。
[0063]
所述的氧化反应的进程可采用本领域常规的检测方式进行监测,如薄层色谱(tlc)、气相色谱(gc)、核磁共振波谱(nmr)或高效液相色谱(hplc)等。
[0064]
所述的氧化反应的时间以所述的氧化反应完全为准,例如0.1~4h,例如1小时。
[0065]
所述的氧化反应的后处理步骤可为本领域该类反应常规的后处理步骤,例如:过滤,柱层析,得到所述的式a所示化合物即可。
[0066]
本发明提供了一种式t所示化合物、其药学上可接受的盐、其溶剂合物或其药学上可接受的盐的溶剂合物在制备nf-κb信号阻断剂中的应用;所述的nf-κb信号阻断剂可为tnf-α介导的nf-κb信号阻断剂;所述的nf-κb信号阻断剂优选为在体外使用的nf-κb信号阻断剂。
[0067]
本发明还提供了一种药物组合物,其包含如上述所示的式t所示化合物、其药学上可接受的盐、其溶剂合物或其药学上可接受的盐的溶剂合物和药用辅料。
[0068]
本发明还提供了一种式t所示化合物、其药学上可接受的盐、其溶剂合物或其药学上可接受的盐的溶剂合物在制备治疗三阴性乳腺癌药物中的应用;所述的三阴性乳腺癌细胞可为mda-mb-231和/或bt-549肿瘤细胞株。
[0069]
术语解释
[0070]
在本说明书的各部分,本发明公开化合物的取代基按照基团种类或范围公开。特别指出,本发明包括这些基团种类和范围的各个成员的每一个独立的次级组合。例如,术语“c1~c4烷基”特指独立公开的甲基、乙基、c3烷基(即丙基,包括正丙基和异丙基)、c4烷基(即丁基,包括正丁基、异丁基、仲丁基和叔丁基)。
[0071]
当所列举的取代基中没有指明其通过哪一个原子连接到化学结构通式中包括但未具体提及的化合物时,这种取代基可以通过其任何原子相键合。取代基和/或其变体的组合只有在这样的组合会产生稳定的化合物的情况下才是被允许的。
[0072]
当所列举的基团中没有明确指明其具有取代基时,这种基团仅指未被取代。例如当“c1~c4烷基”前没有“取代或未取代的”的限定时,仅指“c1~c4烷基”本身或“未取代的c1~c4烷基”。
[0073]
术语“药学上可接受的盐”是指化合物与药学上可接受的(相对无毒、安全、适合于患者使用)酸或碱反应得到的盐。当化合物中含有相对酸性的官能团时,可以通过在合适的惰性溶剂中用足量的药学上可接受的碱与化合物的游离形式接触的方式获得碱加成盐。药学上可接受的碱加成盐包括但不限于钠盐、钾盐、钙盐、铝盐、镁盐、铋盐、铵盐等。当化合物中含有相对碱性的官能团时,可以通过在合适的惰性溶剂中用足量的药学上可接受的酸与化合物的游离形式接触的方式获得酸加成盐。药学上可接受的酸加成盐包括但不限于盐酸盐、硫酸盐、甲磺酸盐等。具体参见handbook of pharmaceutical salts:properties,selection,and use(p.heinrich stahl,2002)。
[0074]
术语“溶剂合物”是指化合物与溶剂(包括但不限于:水、甲醇、乙醇等)结晶后形成的物质。溶剂合物分为化学计量类溶剂合物和非化学计量类溶剂合物。
[0075]
术语“药学上可接受的盐的溶剂合物”是指化合物与药学上可接受的(相对无毒、安全、适合于患者使用)酸或碱、溶剂(包括但不限于:水、甲醇、乙醇等)结合形成的物质,其中,药学上可接受的盐与上文术语“药学上可接受的盐”的含义相同,溶剂为化学计量的或非化学计量的。药学上可接受的盐的溶剂合物包括但不限于盐酸盐一水合物。
[0076]
除非另有规定,本文使用的所有技术术语和科学术语具有要求保护主题所属领域的标准含义。倘若对于某术语存在多个定义,则以本文定义为准。
[0077]
应该理解,在本发明中使用的单数形式,如“一种”,包括复数指代,除非另有规定。此外,术语“包括”是开放性限定并非封闭式,即包括本发明所指明的内容,但并不排除其他方面的内容。
[0078]
除非另有说明,本发明采用质谱、元素分析的传统方法,各步骤和条件可参照本领域常规的操作步骤和条件。
[0079]
除非另有指明,本发明采用分析化学、有机合成化学和光学的标准命名及标准实验室步骤和技术。在某些情况下,标准技术被用于化学合成、化学分析、发光器件性能检测。
[0080]
另外,需要说明的是,除非以其他方式明确指出,在本发明中所采用的描述方式
“…
独立地为”应做广义理解,是指所描述的各个个体之间是相互独立的,可以独立地为相同或不同的具体基团。更详细地,描述方式
“…
独立地为”既可以是指在不同基团中,相同符号之间所表达的具体选项之间互相不影响;也可以表示在相同的基团中,相同符号之间所表达的具体选项之间互相不影响。
[0081]
本领域技术人员可以理解,根据本领域中使用的惯例,本技术描述基团的结构式
中所使用的是指,相应的基团通过该位点与化合物中的其它片段、基团进行连接。
[0082]
在不违背本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。
[0083]
本发明所用试剂和原料均市售可得。
[0084]
本发明的积极进步效果在于:本发明提供了一种去氢中美菊素c衍生物、其制备方法及应用,该化合物既可以阻断nf-κb信号通路的激活,又可以将其应用在制备治疗三阴性乳腺癌药物中,抑制mda-mb-231和bt-549肿瘤细胞株的生长。
具体实施方式
[0085]
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
[0086]
实施例一:去氢中美菊素c衍生物的制备
[0087]
通用操作方法:
[0088]
向去氢木香内酯(6,3.9g,17.1mmol)的二氯甲烷溶液(300ml)中加入二氧化硒(0.94g,8.5mmol)和过氧叔丁醇(5.5m在水中,33.9mmol),搅拌。1小时后缓慢加入百分30%硫代硫酸钠水溶液(150ml)至反应液中。分离有机相,水相用二氯甲烷萃取(150ml
×
2),合并有机相并用饱和食盐水洗涤,无水硫酸钠干燥,并在真空中浓缩。残余物用快速色谱法纯化(pe:etoac=6:4)得到淡黄色固体3-表-中美菊素c(7,2.3g,53%)。光谱数据与文献报道一致(ando,m.et al.org.chem.1989,54,1952)。
[0089]
取3-表-中美菊素c(7,1.23g,5.0mmol)溶于二氯甲烷中(20ml)中,向溶液中分批加入戴斯-马丁过碘烷(2.33g,5.5mmol),室温反应1个小时。tlc监测反应进程,反应结束后加入1m naoh淬灭反应,乙酸乙酯萃取三次(15ml
×
3),并在真空中浓缩。残余物用快速色谱法纯化(pe:etoac=1:1)得到去氢中美菊素c(915mg,75%)。
[0090]
室温下,氮气保护下将吲哚或其他芳杂环(1eq)溶于干燥的二氯甲烷中,向其中加入四氯化锆(0.3eq),反应20min。取去氢中美菊素c(1eq)溶于干燥的二氯甲烷中,冰浴下加入到反应液中,tlc监测反应进程,反应结束后在真空中浓缩。残余物用快速色谱法纯化(pe:etoac=1:1)得到目标产物t。
[0091]
实施例二
[0092]
目标化合物b1的制备:
[0093][0094]
按照通用操作方法,氮气保护下将4-甲氧基吲哚(18mg,0.12mmol)溶于干燥的二
氯甲烷中,向其中加入四氯化锆(8mg,0.04mmol),反应20min。取去氢中美菊素c(8,30mg,0.12mmol)溶于干燥的二氯甲烷中,冰浴下加入到反应液中,tlc监测反应进程,反应结束后在真空中浓缩。残余物用快速色谱法纯化(pe:etoac=1:1)得到目标产物b1(40mg,85%)。1h nmr(500mhz,chloroform-d)δ7.95(s,1h),7.23
–
7.16(m,2h),6.98(s,1h),6.81(dd,j=8.7,2.4hz,1h),6.30(d,j=3.4hz,1h),5.56(d,j=3.0hz,1h),4.95(s,1h),4.62(s,1h),4.06(t,j=8.9hz,1h),3.85(s,3h),3.43(dd,j=14.8,3.3hz,1h),3.22(dd,j=14.7,5.0hz,1h),2.87(m,1h),2.76(t,j=8.5hz,1h),2.61
–
2.49(m,3h),2.38(dt,j=18.5,1.7hz,1h),2.27(m,1h),2.15
–
2.02(m,2h),1.51
–
1.39(m,1h)。
[0095]
目标化合物b2的制备:
[0096][0097]
按照通用操作方法,氮气保护下将吲哚(14mg,0.12mmol)溶于干燥的二氯甲烷中,向其中加入四氯化锆(8mg,0.04mmol),反应20min。取去氢中美菊素c(8,30mg,0.12mmol)溶于干燥的二氯甲烷中,冰浴下加入到反应液中,tlc监测反应进程,反应结束后在真空中浓缩。残余物用快速色谱法纯化(pe:etoac=1:1)得到目标产物b2(37mg,83%)。1h nmr(500mhz,chloroform-d)δ8.03(s,1h),7.77
–
7.71(m,1h),7.32(m,1h),7.13(m,2h),7.04(d,j=2.0hz,1h),6.31(d,j=3.4hz,1h),5.56(d,j=3.0hz,1h),4.95(s,1h),4.63(s,1h),4.05(t,j=9.1hz,1h),3.51
–
3.42(m,1h),3.24(dd,j=14.7,5.4hz,1h),2.87(m,1h),2.76(t,j=8.7hz,1h),2.63
–
2.51(m,3h),2.38(dt,j=18.5,1.8hz,1h),2.26(ddt,j=13.2,5.6,2.7hz,1h),2.09(m,2h),1.51
–
1.39(m,1h)。
[0098]
目标化合物b3的制备:
[0099][0100]
按照通用操作方法,氮气保护下将5-氰基吲哚(17mg,0.12mmol)溶于干燥的二氯甲烷中,向其中加入四氯化锆(8mg,0.04mmol),反应20min。取去氢中美菊素c(8,30mg,0.12mmol)溶于干燥的二氯甲烷中,冰浴下加入到反应液中,tlc监测反应进程,反应结束后在真空中浓缩。残余物用快速色谱法纯化(pe:etoac=1:1)得到目标产物b3(42mg,90%)。1h nmr(500mhz,chloroform-d)δ8.51(s,1h),8.10(d,j=1.2hz,1h),7.37(d,j=1.1hz,2h),7.23(d,j=2.0hz,1h),6.32(d,j=3.4hz,1h),5.58(d,j=3.0hz,1h),4.99(s,1h),4.65(s,1h),4.06(t,j=9.1hz,1h),3.51
–
3.41(m,1h),3.18(dd,j=14.8,5.8hz,1h),2.97
–
2.85(m,2h),2.60(m,2h),2.45(m,1h),2.42(m,1h),2.30(ddt,j=13.3,5.6,2.7hz,1h),2.19
–
2.09(m,2h),1.48(m,1h)。
[0101]
目标化合物b4的制备:
[0102][0103]
按照通用操作方法,氮气保护下将6-氟吲哚(16mg,0.12mmol)溶于干燥的二氯甲烷中,向其中加入四氯化锆(8mg,0.04mmol),反应20min。取去氢中美菊素c(8,30mg,0.12mmol)溶于干燥的二氯甲烷中,冰浴下加入到反应液中,tlc监测反应进程,反应结束后在真空中浓缩。残余物用快速色谱法纯化(pe:etoac=1:1)得到目标产物b4(37mg,82%)。1h nmr(500mhz,chloroform-d)δ8.01(s,1h),7.65(dd,j=8.8,5.4hz,1h),7.02(d,j=2.0hz,1h),6.99(dd,j=9.6,2.3hz,1h),6.87(ddd,j=9.6,8.8,2.3hz,1h),6.31(d,j=3.4hz,1h),5.57(d,j=3.0hz,1h),4.96(s,1h),4.64(s,1h),4.06(t,j=9.1hz,1h),3.44(dd,j=14.7,3.4hz,1h),3.20(dd,j=14.7,5.6hz,1h),2.90(m,1h),2.82(t,j=8.7hz,1h),2.58(m,2h),2.49(m,1h),2.40(dt,j=18.6,1.8hz,1h),2.32
–
2.24(m,1h),2.18
–
2.02(m,2h),1.46(m,1h)。
[0104]
目标化合物b5的制备:
[0105][0106]
按照通用操作方法,氮气保护下将5-氯吲哚(18mg,0.12mmol)溶于干燥的二氯甲烷中,向其中加入四氯化锆(8mg,0.04mmol),反应20min。取去氢中美菊素c(8,30mg,0.12mmol)溶于干燥的二氯甲烷中,冰浴下加入到反应液中,tlc监测反应进程,反应结束后在真空中浓缩。残余物用快速色谱法纯化(pe:etoac=1:1)得到目标产物b5(40mg,85%)。1h nmr(500mhz,chloroform-d)δ8.16(s,1h),7.72(d,j=2.0hz,1h),7.22(d,j=8.5hz,1h),7.12
–
7.05(m,2h),6.32(d,j=3.4hz,1h),5.56(d,j=3.0hz,1h),4.96(s,1h),4.63(s,1h),4.04(t,j=9.1hz,1h),3.39(dd,j=14.8,3.6hz,1h),3.16(dd,j=14.8,5.6hz,1h),2.88(m,1h),2.83
–
2.76(m,1h),2.57(ddd,j=13.1,5.3,2.7hz,2h),2.49
–
2.35(m,2h),2.27(ddt,j=13.3,5.6,2.8hz,1h),2.16
–
2.03(m,2h),1.53
–
1.39(m,1h)。
[0107]
目标化合物b6的制备:
[0108]
[0109]
按照通用操作方法,氮气保护下将2-甲基吲哚(16mg,0.12mmol)溶于干燥的二氯甲烷中,向其中加入四氯化锆(8mg,0.04mmol),反应20min。取去氢中美菊素c(8,30mg,0.12mmol)溶于干燥的二氯甲烷中,冰浴下加入到反应液中,tlc监测反应进程,反应结束后在真空中浓缩。残余物用快速色谱法纯化(pe:etoac=1:1)得到目标产物b6(36mg,81%)。1h nmr(500mhz,chloroform-d)δ7.81(s,1h),7.56
–
7.51(m,1h),7.24
–
7.19(m,1h),7.06(m,2h),6.29(d,j=3.4hz,1h),5.55(d,j=3.1hz,1h),4.92(s,1h),4.62(s,1h),4.01(t,j=9.1hz,1h),3.35(dd,j=14.8,3.6hz,1h),3.16(dd,j=14.8,6.1hz,1h),2.88
–
2.72(m,2h),2.63
–
2.50(m,3h),2.39(s,3h),2.34(m,1h),2.24(m,1h),2.19
–
1.97(m,2h),1.44(m,j=13.3,12.0,5.4hz,1h)。
[0110]
目标化合物b7的制备:
[0111][0112]
按照通用操作方法,氮气保护下将7-氯吲哚(18mg,0.12mmol)溶于干燥的二氯甲烷中,向其中加入四氯化锆(8mg,0.04mmol),反应20min。取去氢中美菊素c(8,30mg,0.12mmol)溶于干燥的二氯甲烷中,冰浴下加入到反应液中,tlc监测反应进程,反应结束后在真空中浓缩。残余物用快速色谱法纯化(pe:etoac=1:1)得到目标产物b7(40mg,85%)。1h nmr(500mhz,chloroform-d)δ8.27(s,1h),7.65(dt,j=8.0,0.8hz,1h),7.15(dd,j=7.7,0.9hz,1h),7.11(d,j=2.3hz,1h),7.04(t,j=7.8hz,1h),6.31(d,j=3.4hz,1h),5.57(d,j=3.0hz,1h),4.96(s,1h),4.63(s,1h),4.05(t,j=9.1hz,1h),3.46(dd,j=14.7,3.5hz,1h),3.22(dd,j=14.7,5.6hz,1h),2.88(m,1h),2.80(t,j=8.7hz,1h),2.68
–
2.51(m,2h),2.51
–
2.35(m,2h),2.27(m,1h),2.16
–
2.04(m,2h),1.52
–
1.40(m,1h)。
[0113]
目标化合物b8的制备:
[0114][0115]
按照通用操作方法,氮气保护下将6-氯吲哚(18mg,0.12mmol)溶于干燥的二氯甲烷中,向其中加入四氯化锆(8mg,0.04mmol),反应20min。取去氢中美菊素c(8,30mg,0.12mmol)溶于干燥的二氯甲烷中,冰浴下加入到反应液中,tlc监测反应进程,反应结束后在真空中浓缩。残余物用快速色谱法纯化(pe:etoac=1:1)得到目标产物b8(40mg,85%)。1h nmr(500mhz,chloroform-d)δ8.05(s,1h),7.65(d,j=8.6hz,1h),7.30(d,j=1.9hz,1h),7.07(dd,j=8.5,1.7hz,1h),7.04(s,1h),6.31(d,j=3.3hz,1h),5.57(d,j=2.9hz,1h),4.96(s,1h),4.63(s,1h),4.05(t,j=9.1hz,1h),3.44(dd,j=14.7,3.4hz,1h),3.20
(dd,j=14.7,5.5hz,1h),2.93
–
2.85(m,1h),2.80(t,j=8.7hz,1h),2.57(m,2h),2.45(m,2h),2.38(s,1h),2.32
–
2.23(m,1h),2.16
–
2.03(m,2h),1.52
–
1.40(m,1h)。
[0116]
目标化合物b9的制备:
[0117][0118]
按照通用操作方法,氮气保护下将4-氯吲哚(18mg,0.12mmol)溶于干燥的二氯甲烷中,向其中加入四氯化锆(8mg,0.04mmol),反应20min。取去氢中美菊素c(8,30mg,0.12mmol)溶于干燥的二氯甲烷中,冰浴下加入到反应液中,tlc监测反应进程,反应结束后在真空中浓缩。残余物用快速色谱法纯化(pe:etoac=1:1)得到目标产物b9(40mg,85%)。1h nmr(500mhz,chloroform-d)δ8.23(s,1h),7.24(dd,j=5.3,3.7hz,1h),7.16(d,j=2.2hz,1h),7.08
–
7.00(m,2h),6.25
–
6.19(m,1h),5.50(d,j=3.1hz,1h),4.96(s,1h),4.77(s,1h),4.02(t,j=8.9hz,1h),3.51
–
3.39(m,2h),3.07(m,1h),2.97
–
2.82(m,2h),2.65
–
2.57(m,1h),2.56
–
2.38(m,3h),2.32
–
2.22(m,1h),1.51
–
1.40(m,1h)。
[0119]
目标化合物b10的制备:
[0120][0121]
按照通用操作方法,氮气保护下将4-甲基吲哚(16mg,0.12mmol)溶于干燥的二氯甲烷中,向其中加入四氯化锆(8mg,0.04mmol),反应20min。取去氢中美菊素c(8,30mg,0.12mmol)溶于干燥的二氯甲烷中,冰浴下加入到反应液中,tlc监测反应进程,反应结束后在真空中浓缩。残余物用快速色谱法纯化(pe:etoac=1:1)得到目标产物b10(37mg,81%)。1h nmr(500mhz,chloroform-d)δ8.25
–
8.19(m,1h),7.14(d,j=8.1hz,1h),7.05
–
6.98(m,2h),6.81(m,1h),6.23(d,j=3.4hz,1h),5.50(d,j=3.0hz,1h),4.95(s,1h),4.73(s,1h),3.97(t,j=8.8hz,1h),3.48(s,1h),3.40
–
3.36(m,2h),3.01
–
2.93(m,1h),2.79(m,2h),2.74(s,3h),2.59
–
2.36(m,4h),2.32
–
2.18(m,1h),2.13(m,1h),1.50
–
1.33(m,1h)。
[0122]
目标化合物b11的制备:
[0123]
[0124]
按照通用操作方法,氮气保护下将5,6-二氟吲哚(18mg,0.12mmol)溶于干燥的二氯甲烷中,向其中加入四氯化锆(8mg,0.04mmol),反应20min。取去氢中美菊素c(8,30mg,0.12mmol)溶于干燥的二氯甲烷中,冰浴下加入到反应液中,tlc监测反应进程,反应结束后在真空中浓缩。残余物用快速色谱法纯化(pe:etoac=1:1)得到目标产物b11(42mg,88%)。1h nmr(500mhz,chloroform-d)δ8.12(s,1h),7.48(dd,j=11.2,7.9hz,1h),7.13
–
7.05(m,2h),6.32(d,j=3.4hz,1h),5.58(d,j=3.1hz,1h),4.97(s,1h),4.63(s,1h),4.05(t,j=9.1hz,1h),3.40(dd,j=14.7,3.5hz,1h),3.13(dd,j=14.8,5.6hz,1h),2.92(m,1h),2.85(t,j=8.7hz,1h),2.57(tdd,j=10.1,4.4,2.1hz,2h),2.51
–
2.38(m,2h),2.29(m,1h),2.18
–
2.06(m,2h),1.53
–
1.40(m,1h)。
[0125]
手性结构确证
[0126][0127]
碳89.2的h和碳52.9的noe相关,碳46.0/39.6/43.9的h noe相关,从而判断构型为:
[0128]
目标化合物b12的制备:
[0129][0130]
按照通用操作方法,氮气保护下将6-氰基吲哚(17mg,0.12mmol)溶于干燥的二氯甲烷中,向其中加入四氯化锆(8mg,0.04mmol),反应20min。取去氢中美菊素c(8,30mg,0.12mmol)溶于干燥的二氯甲烷中,冰浴下加入到反应液中,tlc监测反应进程,反应结束后在真空中浓缩。残余物用快速色谱法纯化(pe:etoac=1:1)得到目标产物b12(41mg,89%)。1h nmr(500mhz,chloroform-d)δ8.60(s,1h),7.81(d,j=8.3hz,1h),7.66(t,j=1.0hz,1h),7.32(dd,j=8.4,1.4hz,1h),7.28(d,j=2.4hz,1h),6.32(d,j=3.4hz,1h),5.59(d,j=3.1hz,1h),4.98(s,1h),4.63(s,1h),4.06(t,j=9.1hz,1h),3.46(dd,j=14.8,3.4hz,1h),3.20(dd,j=14.8,5.7hz,1h),2.91(m,1h),2.84(t,j=9.0hz,1h),2.58(m,2h),2.46
–
2.40(m,1h),2.42
–
2.36(m,1h),2.29(m,1h),2.17
–
2.11(m,1h),1.53
–
1.40(m,1h)。
[0131]
目标化合物b13的制备:
[0132][0133]
按照通用操作方法,氮气保护下将7-甲氧基吲哚(17mg,0.12mmol)溶于干燥的二氯甲烷中,向其中加入四氯化锆(8mg,0.04mmol),反应20min。取去氢中美菊素c(8,30mg,0.12mmol)溶于干燥的二氯甲烷中,冰浴下加入到反应液中,tlc监测反应进程,反应结束后在真空中浓缩。残余物用快速色谱法纯化(pe:etoac=1:1)得到目标产物b13(41mg,88%)。1h nmr(500mhz,chloroform-d)δ8.25(s,1h),7.34(dd,j=8.1,0.8hz,1h),7.06
–
6.97(m,2h),6.61(dd,j=7.7,0.7hz,1h),6.30(d,j=3.5hz,1h),5.55(d,j=3.0hz,1h),4.94(s,1h),4.62(s,1h),4.04(t,j=9.1hz,1h),3.93(s,3h),3.44(dd,j=14.6,3.4hz,1h),3.22(dd,j=14.7,5.3hz,1h),2.85(m,1h),2.77
–
2.66(m,1h),2.61
–
2.41(m,3h),2.37(m,1h),2.25(m,1h),2.13
–
2.01(m,2h),1.50
–
1.36(m,1h)。
[0134]
目标化合物b14的制备:
[0135][0136]
按照通用操作方法,氮气保护下将吲哚-7-甲酸甲酯(21mg,0.12mmol)溶于干燥的二氯甲烷中,向其中加入四氯化锆(8mg,0.04mmol),反应20min。取去氢中美菊素c(8,30mg,0.12mmol)溶于干燥的二氯甲烷中,冰浴下加入到反应液中,tlc监测反应进程,反应结束后在真空中浓缩。残余物用快速色谱法纯化(pe:etoac=1:1)得到目标产物b14(43mg,86%)。1h nmr(500mhz,chloroform-d)δ9.69(s,1h),7.99(d,j=7.9hz,1h),7.85(dd,j=7.6,1.1hz,1h),7.17
–
7.11(m,2h),6.31(d,j=3.4hz,1h),5.56(d,j=3.0hz,1h),4.96(s,1h),4.62(s,1h),4.05(t,j=9.1hz,1h),3.96(s,3h),3.49(dd,j=14.7,3.4hz,1h),3.25(dd,j=14.7,5.5hz,1h),2.88(m,1h),2.78(t,j=8.8hz,1h),2.58(m,2h),2.46(m,1h),2.39(m,1h),2.27(m,1h),2.16
–
2.01(m,2h),1.51
–
1.39(m,1h)。
[0137]
目标化合物b15的制备:
[0138]
[0139]
按照通用操作方法,氮气保护下将5-三氟甲基吲哚(22mg,0.12mmol)溶于干燥的二氯甲烷中,向其中加入四氯化锆(8mg,0.04mmol),反应20min。取去氢中美菊素c(8,30mg,0.12mmol)溶于干燥的二氯甲烷中,冰浴下加入到反应液中,tlc监测反应进程,反应结束后在真空中浓缩。残余物用快速色谱法纯化(pe:etoac=1:1)得到目标产物b15(44mg,84%)。1h nmr(500mhz,chloroform-d)δ8.37(s,1h),8.06(s,1h),7.38(d,j=1.2hz,2h),7.15(d,j=2.3hz,1h),6.32(d,j=3.4hz,1h),5.57(d,j=3.0hz,1h),4.96(s,1h),4.63(s,1h),4.05(t,j=9.2hz,1h),3.49
–
3.42(m,1h),3.23(dd,j=14.7,5.5hz,1h),2.93
–
2.77(m,2h),2.64
–
2.53(m,2h),2.51
–
2.37(m,2h),2.27(m,1h),2.18
–
2.04(m,2h),1.52
–
1.39(m,1h)。
[0140]
目标化合物b16的制备:
[0141][0142]
按照通用操作方法,氮气保护下将5-苄氧基吲哚(27mg,0.12mmol)溶于干燥的二氯甲烷中,向其中加入四氯化锆(8mg,0.04mmol),反应20min。取去氢中美菊素c(8,30mg,0.12mmol)溶于干燥的二氯甲烷中,冰浴下加入到反应液中,tlc监测反应进程,反应结束后在真空中浓缩。残余物用快速色谱法纯化(pe:etoac=1:1)得到目标产物b16(44mg,79%)。1h nmr(500mhz,chloroform-d)δ7.99(s,1h),7.56
–
7.51(m,2h),7.38(dd,j=8.3,6.9hz,2h),7.34
–
7.26(m,2h),7.19(d,j=8.8hz,1h),6.96(d,j=2.2hz,1h),6.90(dd,j=8.7,2.4hz,1h),6.31(d,j=3.4hz,1h),5.55(d,j=3.1hz,1h),5.17
–
5.06(m,2h),4.94(s,1h),4.61(s,1h),4.04(t,j=9.0hz,1h),3.41(dd,j=14.7,3.3hz,1h),3.20(dd,j=14.7,5.2hz,1h),2.82(m,1h),2.71(t,j=8.6hz,1h),2.60
–
2.54(m,1h),2.57
–
2.46(m,2h),2.36(m,1h),2.24(m,1h),2.12
–
1.98(m,2h),1.50
–
1.38(m,1h)。
[0143]
目标化合物b17的制备:
[0144][0145]
按照通用操作方法,氮气保护下将6-三氟甲基吲哚(27mg,0.12mmol)溶于干燥的二氯甲烷中,向其中加入四氯化锆(8mg,0.04mmol),反应20min。取去氢中美菊素c(8,30mg,0.12mmol)溶于干燥的二氯甲烷中,冰浴下加入到反应液中,tlc监测反应进程,反应结束后在真空中浓缩。残余物用快速色谱法纯化(pe:etoac=1:1)得到目标产物b17(43mg,83%)。1h nmr(500mhz,chloroform-d)δ8.50(s,1h),7.82(d,j=8.4hz,1h),7.61
–
7.58(m,1h),7.32
(dd,j=8.5,1.6hz,1h),7.18(d,j=2.3hz,1h),6.32(d,j=3.4hz,1h),5.58(d,j=3.1hz,1h),4.96(s,1h),4.62(s,1h),4.05(t,j=9.1hz,1h),3.45(dd,j=14.7,3.4hz,1h),3.21(dd,j=14.7,5.6hz,1h),2.86(m,1h),2.77(t,j=8.7hz,1h),2.57(tdt,j=7.7,5.3,2.1hz,2h),2.46
–
2.36(m,2h),2.27(ddt,j=13.3,5.5,2.7hz,1h),2.14
–
2.01(m,2h),1.51
–
1.39(m,1h)。
[0146]
目标化合物b18的制备:
[0147][0148]
按照通用操作方法,氮气保护下将苯并呋喃(15mg,0.12mmol)溶于干燥的二氯甲烷中,向其中加入四氯化锆(8mg,0.04mmol),反应20min。取去氢中美菊素c(8,30mg,0.12mmol)溶于干燥的二氯甲烷中,冰浴下加入到反应液中,tlc监测反应进程,反应结束后在真空中浓缩。残余物用快速色谱法纯化(pe:etoac=1:1)得到目标产物b18(19mg,44%)。1h nmr(500mhz,chloroform-d)δ8.50(s,1h),7.82(d,j=8.4hz,1h),7.61
–
7.58(m,1h),7.32(dd,j=8.5,1.6hz,1h),7.18(d,j=2.3hz,1h),6.32(d,j=3.4hz,1h),5.58(d,j=3.1hz,1h),4.96(s,1h),4.62(s,1h),4.05(t,j=9.1hz,1h),3.45(dd,j=14.7,3.4hz,1h),3.21(dd,j=14.7,5.6hz,1h),2.86(ddq,j=11.8,9.2,3.1hz,1h),2.77(t,j=8.7hz,1h),2.57(tdt,j=7.7,5.3,2.1hz,2h),2.46
–
2.36(m,2h),2.27(ddt,j=13.3,5.5,2.7hz,1h),2.14
–
2.01(m,2h),1.51
–
1.39(m,1h)。
[0149]
目标化合物b19的制备:
[0150][0151]
按照通用操作方法,氮气保护下将2-甲基噻吩(12mg,0.12mmol)溶于干燥的二氯甲烷中,向其中加入四氯化锆(8mg,0.04mmol),反应20min。取去氢中美菊素c(8,30mg,0.12mmol)溶于干燥的二氯甲烷中,冰浴下加入到反应液中,tlc监测反应进程,反应结束后在真空中浓缩。残余物用快速色谱法纯化(pe:etoac=1:1)得到目标产物b19(20mg,51%)。1h nmr(500mhz,chloroform-d)δ6.64(d,j=3.3hz,1h),6.55
–
6.50(m,1h),6.29(d,j=3.4hz,1h),5.57(d,j=3.1hz,1h),4.98(s,1h),4.64(s,1h),4.01(t,j=8.9hz,1h),3.47(dd,j=14.8,3.0hz,1h),3.20(dd,j=14.8,5.0hz,1h),2.95(m,2h),2.64
–
2.43(m,4h),2.38(s,3h),2.37
–
2.25(m,2h),2.16(m,1h),1.52
–
1.40(m,1h)。
[0152]
目标化合物b20的制备:
[0153][0154]
按照通用操作方法,氮气保护下将吡咯(8mg,0.12mmol)溶于干燥的二氯甲烷中,向其中加入四氯化锆(8mg,0.04mmol),反应20min。取去氢中美菊素c(8,30mg,0.12mmol)溶于干燥的二氯甲烷中,冰浴下加入到反应液中,tlc监测反应进程,反应结束后在真空中浓缩。残余物用快速色谱法纯化(pe:etoac=1:1)得到目标产物b20(12mg,33%)。1h nmr(500mhz,chloroform-d)δ8.51(s,1h),6.63(m,1h),6.31(d,j=3.5hz,1h),6.06(q,j=2.8hz,1h),5.93(m,1h),5.59(d,j=3.1hz,1h),4.99(s,1h),4.63(d,j=0.8hz,1h),4.03(t,j=9.1hz,1h),3.36(dd,j=15.0,2.9hz,1h),3.03
–
2.90(m,3h),2.61(m,1h),2.56
–
2.44(m,2h),2.40(m,1h),2.36
–
2.26(m,2h),2.24
–
2.11(m,1h),1.45(m,1h)。
[0155]
效果实施例1:化合物对nf-κb细胞萤光素酶表达的影响
[0156]
(1)将需要复苏的细胞从液氮罐内迅速取出,在37℃水浴锅中融化,迅速加入预热的培养基中。1000转/分钟,离心5分钟后将离心管取出,弃去上清液,向离心管内加入新鲜预热的培养基,重悬细胞,然后将细胞悬液加入培养皿中,37℃,5%co2培养。
[0157]
(2)贴壁细胞传代:当细胞长满培养皿80~90%,用0.25%胰酶消化细胞,然后用新的培养基将细胞重悬,将细胞按适当比例传代,约2~4d传代1次。
[0158]
(3)细胞接种及药物处理
[0159]
(a)检测前1天,根据细胞生长速度将细胞按40000细胞每孔接种于96孔细胞板中,每孔接种80μl细胞悬液,37℃,5%co2培养箱,孵育过夜。
[0160]
(b)根据实验要求,每孔加入10μl化合物工作液孵育1小时后,加入10μltnfα(200ng/ml),37℃,5%co2培养箱,避光孵育24小时。
[0161]
(c)结束孵育后,加入bright glo 50μl/孔,在nivo上测定化学发光,计算抑制率。
[0162]
(4)实验结果
[0163]
表1.代表性化合物对nf-κb信号通路的抑制活性
[0164][0165]
效果实施例2:代表化合物对乳腺癌细胞的生长抑制作用
[0166]
(1)细胞培养基配制
[0167]
mda-mb-231:dmem+10%fbs+1%p/s
[0168]
(2)药物配置
[0169]
将化合物用细胞对应培养液稀释成实验设计浓度。
[0170]
(3)细胞增殖实验
[0171]
a.细胞经计数后按照以下密度接种于384孔培养板,每孔40ul。放入37℃,5%co2培养箱过夜培养。
[0172]
b.每孔加入10μl的待测化合物,使化合物终浓度达到excel附件数据layout的浓度。继续培养72h后,每孔加入ctg 100μl。
[0173]
c.室温静置10min,
[0174]
d.在酶标仪上测定各孔化学发光值。
[0175]
(4)实验结果
[0176]
表2.代表化合物对乳腺癌细胞的生长抑制作用
[0177]
化合物mda-mb-231ic
50
(μm)bt549 ic
50
(μm)b33.586.95b53.867.83b112.093.93b121.988.09b132.971.37b184.052.98。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1