一种高纯度叔丁基咔唑的制备方法与流程
1.本发明涉及药物合成技术领域,具体涉及一种高纯度叔丁基咔唑的制备方法。
背景技术:
2.叔丁基咔唑(tert
‑
butyl carbazate),又称为肼基甲酸叔丁酯,叔丁基卡巴酯等。它可用于制备叔丁氧羰基
‑
叠氮化物的原料,而叔丁氧羰基
‑
叠氮化物是引入叔丁氧羰基氨基保护的一种试剂;也可用作合成磺酰肼和羧基肼的试剂;用于与乙烯基卤化物进行钯催化交叉偶联反应,生成n
‑
叔丁氧羰基
‑
n
‑
烯基肼;用于固相多肽合成和α
‑
氨基醛的光学纯度测定的试剂;可与醛发生缩合反应形成腙,腙可作为合成hiv
‑
1蛋白酶抑制剂的中间体。广泛应用于药物、高分子化合物、染料工业。
3.目前文献报道的叔丁基咔唑的制备方法主要有以下几种:
4.方法1:louis a.carpino用叔丁醇钠与羰基硫反应得到(o)
‑
叔丁氧基硫代碳酸钠,再与碘甲烷反应生成甲硫基甲酸叔丁酯,然后与水合肼反应得到叔丁基咔唑。(carpino,louis a.new amino
‑
protecting groups in organic synthesis[j].accounts of chemical research,1973,6(6):191
‑
198.)该合成路线首次制备得到叔丁基咔唑,并应用到了氨基的保护过程中,不过此合成过程,药品价格昂贵,反应时间过长等条件的要求,只能制备小批量的产品,很难实现工业化。
[0005][0006]
方法2:公开号为wo2001040163a1的pct国际申请报道了通过叔丁醇钠与三光气反应得到氯甲酸叔丁酯,再水合肼反应得到叔丁基咔唑。该合成路线反应过程会产生剧毒的光气,容易产生光气泄漏,工艺安全风险较高,对工艺设备的密闭性要求也比较高。
[0007][0008]
方法3:通过氯甲酸苯酯与叔丁醇缩合反应得到苯氧基甲酸叔丁酯,然后与水合肼反应得到叔丁基咔唑,(organic synthesis,coll.vol.5,p.166(1973)vol.44,p.20(1964))。该路线要用到较为昂贵的氯甲酸苯酯,并且产生副产物苯酚难以除去。
[0009][0010]
方法4:通过氯甲酸乙酯与叔丁醇反应生成乙氧基甲酸叔丁酯,然后与水合肼反应得到叔丁基咔唑。该路线所使用的氯甲酸乙酯属于受公安部门管制的剧毒物品。
[0011][0012]
方法5:公开号为cn101823986a的中国发明专利公开了通过二叔丁基二碳酸酯与水合肼直接反应得到叔丁基咔唑的方法,该路线较为简洁,所使用的原料二叔丁基二碳酸酯安全性较好。但是该专利工艺的缺点是所得到的产品纯度不高,产物中有难以去除的副产物叠氮二羧酸二叔丁酯杂质。
[0013]
技术实现要素:
[0014]
本发明要解决的技术问题是:提供一种具有较高的产品纯度、原料简单易得的叔丁基咔唑制备方法。
[0015]
本发明解决上述技术问题的技术方案如下:
[0016]
一种高纯度叔丁基咔唑的制备方法,包括如下步骤:
[0017]
(1)水合肼溶于醇类或醚类溶剂中,添加无机酸或有机酸生成水合肼单酸盐;
[0018]
(2)水合肼单酸盐与二碳酸二叔丁酯缩合,反应结束后,用碱中和,用溶剂萃取,浓缩,用弱极性溶剂析晶,得到叔丁基咔唑产品。
[0019]
优选的,所述步骤(1)中的醇类或醚类溶剂选自甲醇、乙醇、叔丁醇或乙二醇二甲醚。
[0020]
优选的,所述步骤(1)中的水合肼与溶剂的质量比为1:1~5;进一步的,优选的,所述步骤(1)中的水合肼与溶剂的质量比为1:1~2。
[0021]
优选的,所述步骤(1)中的无机酸选自盐酸、硫酸或磷酸中的一种或多种;所述有机酸选自甲酸,醋酸,丙酸,丁酸,草酸,柠檬酸等其中一种或多种。
[0022]
优选的,所述步骤(1)中的水合肼与无机酸或有机酸的摩尔比为1:0.5~1.5;进一步的,所述步骤(1)中的水合肼与无机酸或有机酸的摩尔比为1:1.0~1.2;更进一步的,所述步骤(1)中的水合肼与无机酸或有机酸的摩尔比为1:1.0~1.05。
[0023]
优选的,所述步骤(2)中水合肼单酸盐与二碳酸二叔丁酯的摩尔比为1:0.4~0.8;进一步的,所述水合肼单酸盐与二碳酸二叔丁酯的摩尔比为1:0.5~0.6。所述水合肼单酸盐的摩尔量以步骤(1)中水合肼的投料量计算。
[0024]
所述滴加二碳酸二叔丁酯的温度控制在0~20℃,滴加时间为0.5~4小时;进一步的,所述滴加二碳酸二叔丁酯的温度控制在5~10℃,滴加时间为1~2小时。
[0025]
本发明中化合物的中文命名与结构式有冲突的,以结构式为准;结构式有明显错误的除外。
[0026]
本发明提供的叔丁基咔唑的制备方法能够得到高纯产物(≥99.5%),且兼具高收率(≥90%),操作简单,适于工业化大规模生产。
附图说明
[0027]
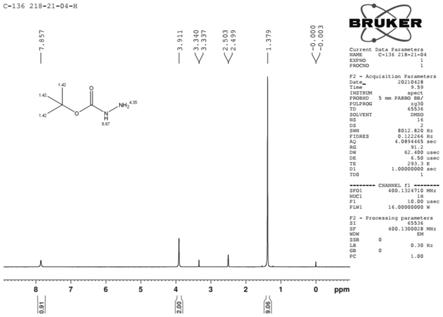
图1为本发明具体实施方式得到的叔丁基咔唑的氢谱图。
具体实施方式
[0028]
以下结合实例说明本发明,但不限制本发明。在本领域内,技术人员对本发明所做的简单替换或改进均属于本发明所保护的技术方案内。
[0029]
实施例1:
[0030]
在5l四口烧瓶加入80%水合肼350g(分子量50.1,5.6mol),甲醇444g,搅拌下,滴加31%盐酸724g(分子量36.5,6.2mol),降温至0~10℃;滴加二碳酸二叔丁酯610g(分子量218.2,2.8mol),约4~6小时滴加完毕。滴加完毕后,0~10℃保温2小时,取样,气相色谱中控至二碳酸二叔丁酯消失,结束反应;将反应液转移至分液漏斗,静置,分层,弃去水相(下层),有机相(上层)浓缩回收溶剂;浓缩液用乙酸乙酯萃取,减压浓缩至干,用石油醚重结晶,得到白色固体339.8g,纯度99.9%(分子量132.2,2.57mol),收率91.8%(以二碳酸二叔丁酯计)。
[0031]
实施例2:
[0032]
在5l四口烧瓶加入80%水合肼350g(分子量50.1,5.6mol),甲醇700g,搅拌下,滴加31%盐酸658g(分子量36.5,5.6mol),降温至0~10℃;滴加二碳酸二叔丁酯733g(分子量218.2,3.4mol),约4~6小时滴加完毕。滴加完毕后,0~10℃保温2小时,取样,气相色谱中控至二碳酸二叔丁酯消失,结束反应;将反应液转移至分液漏斗,静置,分层,弃去水相(下层),有机相(上层)浓缩回收溶剂;浓缩液用乙酸乙酯萃取,减压浓缩至干,用石油醚重结晶,得到白色固体312.4g,纯度99.9%(分子量132.2,2.36mol),收率94.4%(以二碳酸二叔丁酯计)。
[0033]
实施例3:
[0034]
在5l四口烧瓶加入80%水合肼350g(分子量50.1,5.6mol),乙醇500g,搅拌下,滴加31%盐酸790g(分子量36.5,6.7mol),降温至0~10℃;滴加二碳酸二叔丁酯610g(分子量218.2,2.8mol),约4~6小时滴加完毕。滴加完毕后,0~10℃保温2小时,取样,气相色谱中控至二碳酸二叔丁酯消失,结束反应;将反应液转移至分液漏斗,静置,分层,弃去水相(下层),有机相(上层)浓缩回收溶剂;浓缩液用乙酸乙酯萃取,减压浓缩至干,用石油醚重结晶,得到白色固体336.8g,纯度99.8%(分子量132.2,2.54mol),收率90.8%(以二碳酸二叔丁酯计)。
[0035]
实施例4:
[0036]
在5l四口烧瓶加入80%水合肼350g(分子量50.1,5.6mol),甲醇460g,搅拌下,滴
加31%盐酸691g(分子量36.5,5.9mol),降温至0~10℃;滴加二碳酸二叔丁酯672g(分子量218.2,3.1mol),约4~6小时滴加完毕。滴加完毕后,0~10℃保温2小时,取样,气相色谱中控至二碳酸二叔丁酯消失,结束反应;将反应液转移至分液漏斗,静置,分层,弃去水相(下层),有机相(上层)浓缩回收溶剂;浓缩液用乙酸乙酯萃取,减压浓缩至干,用石油醚重结晶,得到白色固体309.8g,纯度99.9%(分子量132.2,2.34mol),收率93.7%(以二碳酸二叔丁酯计)
[0037]
以上所述的仅是本发明的优选实施方式,应当指出,对于本领域的普通技术人员来说,在不脱离本发明创造构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1