一种N-乙酰-L-半胱氨酸合成方法与流程
一种n
‑
乙酰
‑
l
‑
半胱氨酸合成方法
技术领域
1.本发明属于氨基酸合成领域,尤其涉及一种n
‑
乙酰
‑
l
‑
半胱氨酸的合成方法。
背景技术:
2.n
‑
乙酰
‑
l
‑
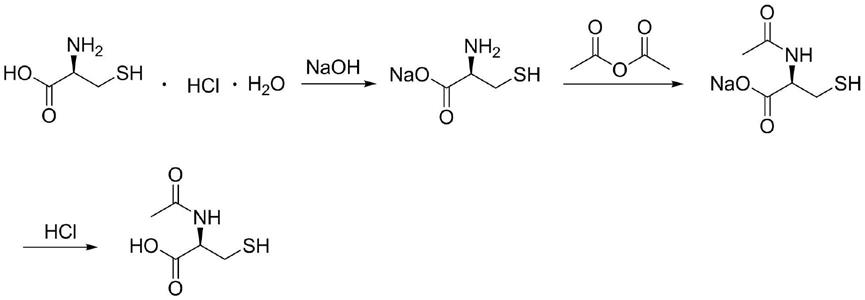
半胱氨酸通过分解粘蛋白复合物、核酸,将痰中的脓性成分及其它粘液和粘液分泌物从粘稠变为稀薄而发挥强烈的粘液溶解,作用适用于大量粘痰阻塞引起的呼吸困难,如手术后的咯痰困难、急性和慢性支气管炎、支气管扩张、肺结核、肺炎、肺气肿等引起的痰液粘稠、咯痰困难、痰阻气管等,还可用于对乙酰氨基酚中毒的解毒,其结构如下所示:
[0003][0004]
目前报道的合成方法主要是用l
‑
半胱氨酸盐酸盐一水物为原料,乙酸酐作为酰化试剂进行合成:比如中国专利cn103102295a、cn104844488a、 cn109096161a等。
[0005]
其中,cn103102295a公开了一种n
‑
乙酰
‑
l
‑
半胱氨酸的生产方法,该方法是l
‑
半胱氨酸盐酸盐一水物与乙酸酐在压力0.4mpa、温度125~135℃下进行酰化反应,该方法对生产设备要求高,能耗大,同时过量的乙酸酐也会导致杂质d大量增加,从而降低反应收率。
[0006]
cn104844488a公开了一种n
‑
乙酰
‑
l
‑
半胱氨酸的生产方法,该方法存在以下缺点:1)解离l
‑
半胱氨酸盐酸盐一水物时没有使用惰性气体保护,会导致底物氧化生成杂质a;2)滴加乙酸酐时温度较高,会导致过乙酰杂质d生成; 3)较低ph值浓缩反应液会导致相关杂质明显增加。该方法收率为质量收率,摩尔收率<60%,可能是相关杂质过多导致收率偏低。
[0007]
cn109096161a公开了一种n
‑
乙酰
‑
l
‑
半胱氨酸的生产方法,该方法以l
‑
半胱氨酸盐酸盐为起始原料,与乙酸酐酰化反应得到n
‑
乙酰
‑
l
‑
半胱氨酸,该方法基本和cn104844488a一致,其收率为质量收率,摩尔收率为62%~74%,并且该方法从母液回收两次粗品后再与第一次粗品合并精制,这不符合药品gmp 生产标准,导致该方法无法用于药品gmp生产。
[0008]
即,现有工艺存在副反应难控制,杂质含量波动较大,实际生产操作繁琐,能耗高等问题,因此,开发一条转化率高、工艺稳定、高收率、低能耗的n
‑ꢀ
乙酰
‑
l
‑
半胱氨酸工业化合成路线具有重大意义。
技术实现要素:
[0009]
本发明的目的就在于提供一种n
‑
乙酰
‑
l
‑
半胱氨酸合成方法,以解决上述问题。
[0010]
为了实现上述目的,本发明采用的技术方案是这样的:
[0011]
一种n
‑
乙酰
‑
l
‑
半胱氨酸的合成方法,包括以下步骤:
[0012]
(1)称取l
‑
半胱氨酸盐酸盐一水物投入容器,氮气保护,再加入纯水,搅拌溶清并降温至
‑
5~10℃;
[0013]
(2)称量氢氧化钠和纯水,配制成氢氧化钠溶液,降温备用;缓慢将配制好的氢氧化钠溶液滴入容器,期间控制内温在
‑
5~10℃,滴毕,保持搅拌5~ 30分钟;
[0014]
(3)称取配比量的乙酸酐,缓慢滴入容器,期间控制内温在
‑
5~10℃,滴毕,保持搅拌5~30分钟;
[0015]
(4)将反应温度升至70~90℃,优选80℃,保温反应1~4小时,优选3 小时,后减压浓缩至干;
[0016]
(5)加入纯水,降温至5~30℃,氮气保护,搅拌均匀;
[0017]
(6)称量盐酸和纯水,配制成2~12n盐酸溶液,备用;
[0018]
(7)滴加配制好的2~12n盐酸溶液,期间有大量白色固体析出,滴毕,降温至
‑
5~10℃,后保温搅拌1~3小时;
[0019]
(8)过滤,滤饼50~90℃鼓风干燥2~6小时,得到n
‑
乙酰
‑
l
‑
半胱氨酸粗品;
[0020]
(9)向容器中依次投入n
‑
乙酰
‑
l
‑
半胱氨酸粗品、纯水,氮气保护,搅拌加热至20~90℃,溶清后保温搅拌5~30分钟;
[0021]
(10)保温过滤,滤液如有固体析出,再次升温至20~90℃溶清;
[0022]
(11)降温至5~30℃,保温析晶2小时,最后缓慢降至
‑
5~10℃,保温析晶 2小时;
[0023]
(12)过滤,烘干,即得n
‑
乙酰
‑
l
‑
半胱氨酸精制品。
[0024]
合成过程为:
[0025][0026]
合成过程中的相关杂质:
[0027][0028]
作为优选的技术方案:所述步骤(1)中,l
‑
半胱氨酸盐酸盐一水物与纯水的重量比为1:2。
[0029]
作为优选的技术方案:所述步骤(2)中,l
‑
半胱氨酸盐酸盐一水物与氢氧化钠固体的摩尔比为1:3,氢氧化钠固体与纯水的重量比为3:17。
[0030]
作为优选的技术方案:所述步骤(3)中,l
‑
半胱氨酸盐酸盐一水物与乙酸酐的摩尔比为1:1。
[0031]
作为优选的技术方案:所述步骤(5)中,l
‑
半胱氨酸盐酸盐一水物与纯水的重量比为1:1。
[0032]
作为优选的技术方案:所述步骤(6)中,精盐酸与纯水的重量比为1:1。
[0033]
作为优选的技术方案:所述步骤(9)中,l
‑
半胱氨酸盐酸盐一水物与纯水的重量比为1:1。
[0034]
作为优选的技术方案:所述步骤(12)中,过滤后,放入烘盘,控制烘箱内温70℃,鼓风干燥4小时,烘料结束,放空,自然降温至30℃以下,收料,称重,得到n
‑
乙酰
‑
l
‑
半胱氨酸精制品。
[0035]
上述的优选方案,副反应更少工艺更稳定、成本更低。
[0036]
与现有技术相比,本发明的优点在于:
[0037]
(1)用碱解离l
‑
半胱氨酸盐酸盐一水物时,通过氮气保护能明显减少药典杂质a的生成;
[0038]
(2)
‑
5~10℃滴加乙酸酐能明显减少药典杂质d的生成;
[0039]
(3)80℃酰化反应3小时反应转化率提高至97%以上并降低药典杂质b、c、 d;
[0040]
(4)调酸后不用浓缩溶剂即能过滤得到产物,高效节能,同时避免酸性条件下产物受热分解生成相关杂质;
[0041]
(5)不用回收粗品母液即有较高摩尔收率,可用于药品gmp生产。
具体实施方式
[0042]
实施例1
[0043]
将150kg l
‑
半胱氨酸盐酸盐一水物投入反应釜,再抽入纯水300kg,氮气保护,搅拌降温至
‑
5℃;加入40%氢氧化钠溶液256kg,滴毕,保温
‑
5℃搅拌 30min;滴加醋酸酐90kg,滴毕,保温
‑
5℃搅拌30min;升至内温90℃反应4 小时,后减压浓缩至干;加入纯水150kg,滴加9n盐酸247kg,滴毕降温至
‑
5℃析晶3小时;过滤烘干,得到n
‑
乙酰
‑
l
‑
半胱氨酸粗品124.3kg;
[0044]
将124.3kg n
‑
乙酰
‑
l
‑
半胱氨酸粗品投入反应釜,再抽入纯水124kg,氮气保护,升温至90℃溶清后降至
‑
5℃析晶3小时;过滤烘干,得到n
‑
乙酰
‑
l
‑
半胱氨酸精制品111.9kg。
[0045]
实施例2
[0046]
将600kg l
‑
半胱氨酸盐酸盐一水物投入反应釜,再抽入纯水1200kg,氮气保护,搅拌降温至5℃;加入15%氢氧化钠溶液2732kg,滴毕,保温5℃搅拌 5min;滴加醋酸酐340kg,滴毕,保温5℃搅拌5min;升至内温70℃反应1小时,后减压浓缩至干;加入纯水600kg,滴加6n盐酸1460kg,滴毕降温至5℃析晶1小时;过滤烘干,得到n
‑
乙酰
‑
l
‑
半胱氨酸粗品499.8kg;
[0047]
将499.8kg n
‑
乙酰
‑
l
‑
半胱氨酸粗品投入反应釜,再抽入纯水500kg,氮气保护,升温至70℃溶清后降至5℃析晶1小时;过滤烘干,得到n
‑
乙酰
‑
l
‑
半胱氨酸精制品449.9kg。
[0048]
实施例3
[0049]
将500kg l
‑
半胱氨酸盐酸盐一水物投入反应釜,再抽入纯水1000kg,氮气保护,搅拌降温至10℃;加入30%氢氧化钠溶液1140kg,滴毕,保温10℃搅拌20min;滴加醋酸酐300kg,滴毕,保温10℃搅拌20min;升至内温80℃反应3小时,后减压浓缩至干;加入纯水500kg,滴加3n盐酸2446kg,滴毕降温至10℃析晶2小时;过滤烘干,得到n
‑
乙酰
‑
l
‑
半胱氨酸粗品418.6kg;
[0050]
将418.6kg n
‑
乙酰
‑
l
‑
半胱氨酸粗品投入反应釜,再抽入纯水420kg,氮气保护,升温至50℃溶清后降至10℃析晶2小时;过滤烘干,得到n
‑
乙酰
‑
l
‑ꢀ
半胱氨酸精制品376.7kg。
[0051]
试验例
[0052]
对实施例1~3进行检测,包括相关物质、产品收率、n
‑
乙酰
‑
l
‑
半胱氨酸纯度等,以cn104844488a(对比例1)、cn109096161a(对比例2)为对比例,其中相关物质(hplc)的检测结果见表1,
[0053]
表1所得数据的检测条件:
[0054]
仪器型号为岛津lc
‑
20ad自动进样高效液相系统;
[0055]
spd
‑
20a检测器,色谱柱为安捷伦c18150mm*4.6mm,5um;
[0056]
检测波长:214nm流速:1.0ml/min进样量:10微升;
[0057]
柱温:40℃进样浓度:1mg/ml运行时间:15min;
[0058]
流动相a:取4ml磷酸和1000ml纯水,混合均匀;
[0059]
流动相b:取4ml磷酸和1000ml甲醇,混合均匀;
[0060]
梯度洗脱的洗脱程序:
[0061][0062]
配样试剂:准确称量0.125g依地酸二钠(edta
‑
na2)和25g氢氧化钠于 500ml容量瓶中,加入300ml纯水溶解固体,冷却至室温,再用纯水稀释至刻度,混合均匀作为储备稀释液,取5.0ml储备稀释液至1000ml容量瓶中,用纯水稀释至刻度,混合均匀。
[0063]
上述检测条件的杂质检测极限0.001%;
[0064]
表1实施例和对比例的检测结果
[0065]
[0066][0067]
表1中,“纯度”是指“n
‑
乙酰
‑
l
‑
半胱氨酸纯度%”;“单杂”是指“最大单个未知杂质”,“标准”是指“原料药gmp质量标准”。
[0068]
结果表明,实施例1~3的相关物质在达到原料药gmp质量标准的基础上,明显优于cn104844488a、cn109096161b,其中,药典杂质a几乎没有,杂质d 未检出。
[0069]
表2实施例和对比例转化率、质量收率、摩尔收率
[0070][0071]
从表2可以看出,实施例1~3产品质量收率、摩尔收率均高于 cn104844488a、cn109096161a。
[0072]
对比例3
[0073]
本对比例与实施例3相比,仅将滴加乙酸酐温度调整为15~20℃,其余与实施例3相同,结果如表3。
[0074]
对比例4
[0075]
本对比例与实施例3相比,仅将滴加乙酸酐温度调整为
‑
10℃,其余与实施例3相同,结果如表3。
[0076]
对比例5
[0077]
本对比例与实施例3相比,仅将“乙酸酐滴毕后,升温至80℃反应3小时”调整为“乙酸酐滴毕后,升温至100℃反应3小时”,其余与实施例3相同,结果如表3。
[0078]
对比例6
[0079]
本对比例与实施例3相比,仅将“调酸后降温析晶过滤”调整为“调酸后 70~80℃浓缩溶剂后再结晶过滤”,其余与实施例3相同,结果如表3。
[0080]
对比例7
[0081]
本对比例与实施例3相比,不用氮气保护,其余与实施例3相同,结果如表3。
[0082]
表3对比例3
‑
7的相关结果:
[0083][0084]
表3中,“纯度”是指“n
‑
乙酰
‑
l
‑
半胱氨酸纯度%”;“单杂”是指“最大单个未知杂质”,“标准”是指“原料药gmp质量标准”。
[0085]
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1