创制高产大豆的方法
1.本发明涉及植物生物技术领域,具体涉及同时基因编辑修饰gmjagged1
‑
1和gmjagged1
‑
2获得高产大豆的方法。
背景技术:
2.大豆通称黄豆,是重要粮食作物和油料作物之一。产量相关的性状是决定大豆高产的重要因素,例如增加单荚粒数可以导致单株籽粒数增加,继而单株籽粒重增加,最后达成大豆产量增加。因而寻找能够改良产量相关的性状,增加大豆产量的基因具有重要意义。
3.大豆的ln位点控制大豆叶片形态及单荚粒数。携带ln位点的大豆材料往往表现为窄叶片且以三粒荚为主,具有四粒荚。图位克隆表明位于大豆第20号染色体上的gmjagged1
‑
2单碱基突变是控制该位点的主效基因(fang,et al.,2013;jeong et al.,2012)。育种实践证明,将ln位点引入不包含该位点的大豆材料中可以创制新的大豆品种,产量可以增加8%到10%(liu et al.,2020)。gmjagged1
‑
1位于大豆第10号染色体上,与gmjagged1
‑
2高度同源。迄今未有报道有同时突变gmjagged1
‑
1和gmjagged1
‑
2的大豆材料和相关表型,因而通过创制同时突变gmjagged1
‑
1和gmjagged1
‑
2的大豆材料改善大豆的产量具有深远的现实意义。
4.参考文献
5.fang,c.,li,w.,li,g.,wang,z.,zhou,z.,ma,y.,shen,y.,li,c.,wu,y.,zhu,b.,yang,w.and tian,z.(2013)cloning of ln gene through combined approach of map
‑
based cloning and association study in soybean.j genet genomics 40,93
‑
96.
6.jeong,n.,suh,s.j.,kim,m.
‑
h.,lee,s.,moon,j.
‑
k.,kim,h.s.andjeong,s.
‑
c.(2012)ln is a key regulator of leaflet shape and number of seeds per pod in soybean.the plant cell 24,4807
‑
4818.
7.liu,s.,zhang,m.,feng,f.and tian,z.(2020)toward a"green revolution"for soybean.molecular plant 13,688
‑
697.
技术实现要素:
8.本发明所要解决的技术问题是如何调控大豆的产量或如何调控大豆的籽粒数和/或籽粒重。
9.为了解决上述技术问题,本发明首先提供了调控蛋白质a和蛋白质b活性或含量的物质或调控所述蛋白质a的编码基因和所述蛋白质b的编码基因的表达物质的应用,所述应用为下述任一种:
10.p1、在调控大豆产量或提高大豆产量中的应用;
11.p2、在调控大豆籽粒数或增加大豆籽粒数中的应用;
12.p3、在调控大豆籽粒重或增加大豆籽粒重中的应用;
13.p4、在大豆育种中的应用;
14.所述蛋白质a可为如下a1)、a2)或a3)的蛋白质:
15.a1)氨基酸序列是序列表中序列3的蛋白质;
16.a2)将a1)所示的氨基酸序列经过一个以上氨基酸残基的取代和/或缺失和/或添加得到的且具有相同功能的由a1)衍生的或与a1)所示的蛋白质具有80%以上的同一性且具有相同功能的蛋白质;
17.a3)在a1)、a2)或a3)的n末端或/和c末端连接蛋白标签得到的融合蛋白质;
18.所述蛋白质b可为如下b1)、b2)或b3)的蛋白质:
19.b1)氨基酸序列是序列表中序列4的蛋白质;
20.b2)将b1)所示的氨基酸序列经过一个以上氨基酸残基的取代和/或缺失和/或添加得到的且具有相同功能的由b1)衍生的或与b1)所示的蛋白质具有80%以上的同一性且具有相同功能的蛋白质;
21.b3)在b1)、b2)或b3)的n末端或/和c末端连接蛋白标签得到的融合蛋白质。
22.上述应用中,所述蛋白质a、所述蛋白质b均可来源于大豆。
23.上述应用中,序列表中序列3由257个氨基酸残基组成,序列表中序列4由256个氨基酸残基组成。
24.上文所述一个以上氨基酸残基具体可为十个以内的氨基酸残基。
25.上述应用中,所述80%以上的同一性可为至少80%、81%、82%、83%、84%、85%、86%、87%、88%、89%、90%、91%、92%、93%、94%、95%、96%、98%或99%的同一性。
26.上述应用中,所述蛋白质a的编码基因可为如下a1)或a2)或a3)所示的dna分子:
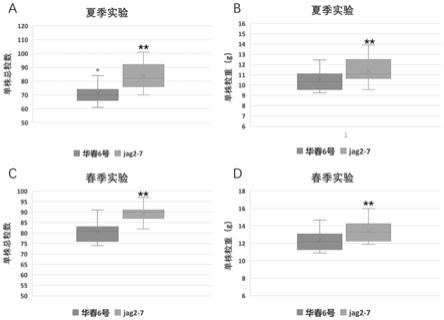
27.a1)编码序列是序列表中序列1所示的dna分子;
28.a2)与a1)限定的核苷酸序列具有90%或90%以上同一性,且编码上文所述蛋白质a的dna分子;
29.a3)在严格条件下与a1)或a3)限定的核苷酸序列杂交,且编码上文所述蛋白质a的dna分子。
30.上述应用中,所述蛋白质b的编码基因可为如下b1)或b2)或b3)所示的dna分子:
31.b1)编码序列是序列表中序列2所示的dna分子;
32.b2)与b1)限定的核苷酸序列具有90%或90%以上同一性,且编码上文所述蛋白质b的dna分子;
33.b3)在严格条件下与b1)或b3)限定的核苷酸序列杂交,且编码上文所述蛋白质b的dna分子。
34.上述应用中,所述调控所述蛋白质a的编码基因和蛋白质b的编码基因表达的物质可为进行如下6种调控中至少一种调控的物质:1)在所述基因转录水平上进行的调控;2)在所述基因转录后进行的调控(也就是对所述基因的初级转录物的剪接或加工进行的调控);3)对所述基因的rna转运进行的调控(也就是对所述基因的mrna由细胞核向细胞质转运进行的调控);4)对所述基因的翻译进行的调控;5)对所述基因的mrna降解进行的调控;6)对所述基因的翻译后的调控(也就是对所述基因翻译的蛋白质的活性进行调控)。
35.上述应用中,调控蛋白质a和蛋白质b活性或含量的物质可为敲除所述蛋白质a的编码基因和蛋白质b的编码基因的物质和/或调控所述蛋白质a的编码基因和蛋白质b的编码基因表达的物质。
36.上述应用中,所述调控所述蛋白质a的编码基因和蛋白质b的编码基因表达的物质可为抑制或降低所述基因表达,所述抑制或降低所述基因表达可通过基因敲除实现或通过基因沉默实现。
37.所述基因敲除(geneknockout)是指通过同源重组使特定靶基因失活的现象。基因敲除是通过dna序列的改变使特定靶基因失活。
38.所述基因沉默是指在不损伤原有dna的情况下使基因不表达或低表达的现象。基因沉默以不改变dna序列为前提,使基因不表达或低表达。基因沉默可发生在两种水平上,一种是由于dna甲基化、异染色质化以及位置效应等引起的转录水平的基因沉默,另一种是转录后基因沉默,即在基因转录后的水平上通过对靶标rna进行特异性抑制而使基因失活,包括反义rna、共抑制(co
‑
suppression)、基因压抑(quelling)、rna干扰(rnai)和微小rna(mirna)介导的翻译抑制等。
39.上述应用中,所述调控所述蛋白质a的编码基因和蛋白质b的编码基因表达的物质可为抑制或降低所述基因表达的试剂。所述抑制或降低所述基因表达的试剂可为敲除所述基因的试剂如通过同源重组敲除所述基因的试剂或通过crispr
‑
cas9敲除所述基因的试剂。所述抑制或降低所述基因表达的试剂可以包含靶向所述基因的多核苷酸,例如sirna、shrna、sgrna、mirna或反义rna。
40.上述应用中,调控蛋白质a和蛋白质b活性或含量的物质或调控所述蛋白质a的编码基因和所述蛋白质b的编码基因的表达物质,可为如下c1)
‑
c4)任一种物质:
41.c1)抑制或降低上文所述蛋白质a编码基因和蛋白质b编码基因表达的核酸分子;
42.c2)含有c1)所述核酸分子的表达盒;
43.c3)含有c1)所述核酸分子的重组载体、或含有c2)所述表达盒的重组载体;
44.c4)含有c1)所述核酸分子的重组微生物、或含有c2)所述表达盒的重组微生物、或含有c3)所述重组载体的重组微生物。
45.c1)所述的核酸分子可为表达同时靶向上文所述所述蛋白质a编码基因和蛋白质b编码基因表达的sgrna或表达所述sgrna的dna分子。
46.所述sgrna为名称为sgrna1的sgrna和名称为sgrna2的sgrna,sgrna1的靶序列为序列1的第405
‑
426位(同序列表中序列2的第221
‑
242位),所述sgrna2的靶序列为序列表序列1的第592
‑
613位(序列表中序列2的第387
‑
408位)所示。
47.术语“同一性”指与天然核酸序列的序列相似性。同一性可以用肉眼或计算机软件进行评价。使用计算机软件,两个或多个序列之间的同一性可以用百分比(%)表示,其可以用来评价相关序列之间的同一性。所述具有90%或90%以上同一性可为至少具有90%、至少具有95%、至少具有96%、至少具有97%、至少具有98%或至少具有99%的同一性。
48.为了解决上述技术问题,本发明还提供了一种增加大豆产量和/或增加大豆籽粒数和/或增加大豆籽粒重的方法,包括通过抑制或降低大豆基因组中上文所述的蛋白质a编码基因和蛋白质b编码基因的表达量来增加大豆产量和/或增加大豆籽粒数和/或增加大豆籽粒重。
49.上述降低或抑制大豆中上文所述蛋白质a编码基因和蛋白质b编码基因的表达量可采用现有技术中的任何方式实现,以使基因产生缺失突变、插入突变或碱基变换突变,进而实现基因功能降低或丧失,具体可为化学诱变、物理诱变、rnai、基因组定点编辑或同源
重组等。
50.上述基因组定点编辑方法中,可采用锌指核酸酶(zinc finger nuclease,zfn)技术、类转录激活因子效应物核酸酶(transcription activator
‑
like effector nuclease,talen)技术或成簇的规律间隔的短回文重复序列及其相关系统(clustered regularly interspaced short palindromic repeats/crispr associated,crispr/cas9 system)技术,以及其它能实现基因组定点编辑的技术。无论采取哪种方法,既可对上文所述蛋白的整个编码基因作为靶标,又可将调控上文所述蛋白编码基因表达的各个元件作为靶标,只要能实现基因功能丧失或降低即可。如可以将上文所述蛋白的编码基因的外显子或5’utr等作为靶标。
51.上文所述方法可包括向所述大豆中导入降低或抑制上文所述的蛋白质a和蛋白质b的活性或所述蛋白质a编码基因和蛋白质b编码基因表达的物质。所述降低或抑制上文所述的蛋白质a和蛋白质b的活性或所述蛋白质a编码基因和蛋白质b编码基因表达的物质可为如下c1)
‑
c4)任一种物质:
52.c1)抑制或降低上文所述蛋白质a编码基因和蛋白质b编码基因表达的核酸分子;
53.c2)含有c1)所述核酸分子的表达盒;
54.c3)含有c1)所述核酸分子的重组载体、或含有c2)所述表达盒的重组载体;
55.c4)含有c1)所述核酸分子的重组微生物、或含有c2)所述表达盒的重组微生物、或含有c3)所述重组载体的重组微生物。
56.c1)所述的核酸分子为靶向上文所述蛋白质a编码基因和上文所述蛋白质b编码基因的sgrna或表达所述sgrna的dna分子。
57.所述sgrna为名称为sgrna1的sgrna和名称为sgrna2的sgrna,sgrna1的靶序列为序列1的第405
‑
426位(同序列表中序列2的第221
‑
242位),所述sgrna2的靶序列为序列表序列1的第592
‑
613位(序列表中序列2的第387
‑
408位)所示。
58.上文所述抑制或降低大豆基因组中所述的蛋白质a编码基因和所述蛋白质b编码基因的表达量为将大豆基因组中的序列1所示的所述蛋白质a编码基因替换为gmjagged1
‑
1基因,并将序列2所示的所述蛋白质b编码基因替换为gmjagged1
‑
2基因,所述gmjagged1
‑
1基因是将序列表中序列1的第419
‑
420位的核苷酸ca和第597
‑
560位的核苷酸gagt均缺失,保持序列表中序列1的其它核苷酸不变得到的dna分子;所述gmjagged1
‑
2基因是将序列表中序列2的第235
‑
238位的核苷酸catg和第393位的核苷酸a均缺失,保持序列表中序列2的其它核苷酸不变得到的dna分子。
59.本发明公开了对大豆gmjagged1
‑
1基因和gmjagged1
‑
2基因同时进行编辑修饰,获得高产大豆的方法。具体利用crispr/cas9系统对出发大豆中gmjagged1
‑
1和gmjagged1
‑
2基因进行同时编辑,且使所述gmjagged1
‑
1和gmjagged1
‑
2基因同时发生突变导致翻译蛋白提前终止,得到转基因大豆;实现出发大豆中gmjagged1
‑
1和gmjagged1
‑
2基因同时基因编辑。本发明利用crispr/cas9介导的基因编辑技术对大豆gmjagged1
‑
1和gmjagged1
‑
2基因进行特定靶点的定点敲除,获得产量较对照提高8%以上的高产大豆突变体材料,为高产大豆品种选育提供新材料。
附图说明
60.图1为突变体jag2
‑
7的gmjagged1
‑
1基因和gmjagged1
‑
2基因在两个靶点处的突变类型。
61.图2为突变体(jag2
‑
1)和野生型(华春6号)在夏季和春季的单株粒数和单株粒重统计。其中,图2的a为突变体(jag2
‑
1)和野生型(华春6号)在夏季的单株粒数统计;图2的b为突变体(jag2
‑
1)和野生型(华春6号)在夏季的单株粒重统计;图2的c为突变体(jag2
‑
1)和野生型(华春6号)在春季的单株粒数统计;图2的d为突变体(jag2
‑
1)和野生型(华春6号)在春季的单株粒重统计。显著性利用t
‑
ttest计算,**代表在0.01水平显著。
具体实施方式
62.下面结合具体实施方式对本发明进行进一步的详细描述,给出的实施例仅为了阐明本发明,而不是为了限制本发明的范围。以下提供的实施例可作为本技术领域普通技术人员进行进一步改进的指南,并不以任何方式构成对本发明的限制。
63.下述实施方法和实施例中所使用的术语,除非特殊说明,一般具有本领域的普通技术人员通常理解的含义。
64.下述实施例中的实验方法,如无特殊说明,均为常规方法,按照本领域内的文献所描述的技术或条件或者按照产品说明书进行。下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
65.以下实施例中,大豆品种华春6号是由华南农业大学农学院选育而成的大豆品种,2009年7月28日经第二届国家农作物品种审定委员会第三次会议审定通过,审定编号为国审豆2009012。
66.以下实施例中,大豆crispr/cas9载体pges201记载于非专利文献“mengyan bai1,juehui yuan1,huaqin kuang,pingping gong,suning li,zhihui zhang,bo liu,jiafeng sun,maoxiang yang,lan yang,dong wang,shikui song,yuefeng guan.(2020)generation of a multiplex mutagenesis population via pooled crispr
‑
cas9 in soya bean.plant biotechnology journal 18,721
‑
731”,公众可以从华南农业大学获得,以重复本技术实验,不可作为其它用途使用。
67.实施例1
68.一、构建同时敲除大豆gmjagged1
‑
1和gmjagged1
‑
2基因的crispr/cas9载体
69.1、sgrna的获得
70.从phytozome数据库获得大豆gmjagged1
‑
1和gmjagged1
‑
2基因的基因组序列。gmjagged1
‑
1基因位于大豆第10号染色体上(49647129
‑
49649853),基因组全长2725bp,如序列表中序列1所示,编码氨基酸序列如序列表中序列3所示的蛋白质;gmjagged1
‑
2基因位于大豆第20号染色体上(35827672
‑
35830107),基因组全长2436bp,如序列表中序列2所示,编码氨基酸序列如序列表中序列4所示的蛋白质。利用crispr
‑
p(http://crispr.hzau.edu.cn/crispr2/)工具进行靶点设计并选择可以同时编辑gmjagged1
‑
1和gmjagged1
‑
2基因的公共靶点。最终获得靶点1和靶点2:
71.靶点1:5
’‑
ctctctgtcggtaccatgtagg
‑3’
(位于序列表中序列1的第405
‑
426位,位于序列表中序列2的第221
‑
242位);
72.靶点2:5
’‑
ccgatgagtactctagggatgg
‑3’
(位于序列表中序列1的第592
‑
613位,位于序列表中序列2的第387
‑
408位)。
73.设计包含两个靶点信息的引物:
74.根据靶点1设计出的引物序列如下:
75.sgrna1 forwardprimer:5
’‑
ggattgctctctgtcggtaccatgt
‑3’
;
76.sgrna1 reverseprimer:5
’‑
aaacacatggtaccgacagagagca
‑3’
。
77.根据靶点2设计出的引物序列如下:
78.sgrna2 forwardprimer:5
’‑
ggattgccatctctagagtactcat
‑3’
;
79.sgrna2 reverseprimer:5
’‑
aaacatgagtactctagagatggca
‑3’
。
80.合成上述设计的引物,用水稀释至10μm后进行退火,得到带粘性末端的双链dna片段sgrna,具体为:
81.(1)sgrna1
82.将引物sgrna1 forward primer和sgrna1 reverse primer退火获得带粘性末端的双链dna片段sgrna1。
83.反应体系为25μl体系:sgrna1 forward primer和sgrna1 reverse primer各5μl,水15μl。
84.反应条件:96℃5min,0.1℃/s退火至12度且保持5min后完成退火,得到带有粘性末端的sgrna1。
85.(2)sgrna2
86.将引物sgrna2 forward primer和sgrna2 reverse primer退火获得带粘性末端的双链dna片段sgrna2。
87.反应体系均为25μl体系:sgrna2 forward primer和sgrna2 reverse primer各5μl,水15μl。
88.反应条件:96℃5min,0.1℃/s退火至12度且保持5min后完成退火,得到带有粘性末端的sgrna2。
89.2、表达sgrna的载体的制备
90.利用bsai酶切大豆crispr/cas9载体pges201载体1μg,回收得到pges201线性化载体,备用。
91.将步骤1获得的带有粘性末端的sgrna1和带有粘性末端的sgrna2,以及上述pges201线性化载体,通过表1的体系连接,完成最终载体crispr/cas9
‑
sgrna1
‑
sgrna2的构建。crispr/cas9
‑
sgrna1
‑
sgrna2表达靶向靶点1的sgrna1和靶向靶点2的sgrna2。
92.表1
93.sgrna11μlsgrna21μlpges201线性化载体2μl10
×
t4 dna连接酶buffer2μlt4 dna连接酶1μl无菌水13μl
94.二、大豆gmjagged1
‑
1和gmjagged1
‑
2双突变体的获得
95.1、将步骤一中获得的crispr/cas9
‑
sgrna1
‑
sgrna2载体转入大肠杆菌感受态dh5α,并涂布于lb+kan的固体培养基上,挑单克隆提取质粒并进行测序,测序引物为sgrna1 forward primer和sgrna2 reverse primer。
96.sgrna1 forward primer:5
’‑
ggattgctctctgtcggtaccatgt
‑3’
;
97.sgrna2 reverse primer:5
’‑
aaacatgagtactctagagatggca
‑3’
。
98.测序比对完成获得正确的crispr/cas9
‑
sgrna1
‑
sgrna2载体质粒。
99.2、用电击转化法将重组质粒crispr/cas9
‑
sgrna1
‑
sgrna2转入根癌农杆菌eha105,提取质粒进行测序验证,将测序验证正确(含有sgrna1编码序列和sgrna2编码序列)的重组菌株分别命名eha105
‑
crispr/cas9
‑
sgrna1
‑
sgrna2。
100.3、华春6号大豆的遗传转化:
101.以大豆栽培品种华春6号(公众可从华南农业大学农学院国家大豆改良中心广东分中心获得)为受体材料,通过子叶节法进行稳定转化,方法参考文献“li s,cong y,liu y,wang t,shuai q,chen n,gai j,li y.optimization of agrobacterium
‑
mediated transformation in soybean.front plant sci.2017 feb24;8:246.doi:10.3389/fpls.2017.00246.pmid:28286512;pmcid:pmc5323423.”,根据大豆转化方法的参考文献做简要修改如下:将农杆菌侵染好的大豆放置在脱分化培养基上培养5天,然后转移至再分化培养基上培养14天,然后再转移至含basta的抗性再分化培养基上培养14天,然后再转移至含植物激素和basta的抗性再分化培养基上培养28天,最后转移至根诱导培养基上培养。获得的植株转移到土壤中生长,并用1:1000稀释的除草剂basta喷施筛选阳性苗,得到t0阳性大豆植株。
102.4、gmjagged1
‑
1和gmjagged1
‑
2双突变体筛选和鉴定
103.提取t0阳性大豆植株的叶片的dna作为模板进行pcr分子检测,以野生型大豆华春6号为对照。设计引物包含靶点sgrna1和sgrna2在内的pcr引物,进行pcr扩增,并将扩增产物进行测序。其中扩增gmjagged1
‑
1的引物为:gmjagged1
‑
1f(作为引物1)和gmjagged1
‑
1r(作为引物2);扩增gmjagged1
‑
2的引物为:gmjagged1
‑
2f(作为引物1)和gmjagged1
‑
2r(作为引物2)。
104.gmjagged1
‑
1f:5
’‑
tcaaccaaaccagacgaagc
‑3’
;
105.gmjagged1
‑
1r:5
’‑
aaccacggcagtgttcaact
‑3’
。
106.gmjagged1
‑
2f:5
’‑
aaagtcacgagacccactgtc
‑3’
;
107.gmjagged1
‑
2r:5
’‑
tgaaaggcttcccatggtgg
‑3’
。
108.pcr试剂体系如下表2:
109.表2
[0110]2×
phanta max buffer25μldntp mix(10mm)1μlsuper
‑
fidelity dna polymerase1μl引物11μl引物21μl无菌水21μl
[0111]
pcr反应体系为:95℃3min;95℃30sec,58℃30sec,72℃2min,35个循环;72℃
5min。
[0112]
pcr产物送公司测序验证。测序结果表明在靶点位置附近出现套峰的植株为杂合编辑植株,命名为t0代gmjagged1
‑
1和gmjagged1
‑
2双编辑大豆。
[0113]
以t0代gmjagged1
‑
1和gmjagged1
‑
2双编辑大豆自交后收获的t1代种子种植得到t1代植株,采用上述步骤4中的pcr体系扩增并测序,获得了一株在gmjagged1
‑
1和gmjagged1
‑
2基因的sgrna1和sgrna2位置均发生编辑的大豆植株jag2
‑
7(t1代),具体见图1:该材料的gmjagged1
‑
1基因在靶点1位置上缺失序列表序列1第419
‑
420位的核苷酸ca,共缺失2bp,在靶点2位置上,缺失序列表序列1第597
‑
560位的核苷酸gagt,共缺失4bp,导致cds起始密码子缺失且第二个外显子移码突变,最终致使gmjagged1
‑
1功能丧失,产生gmjagged1
‑
1基因,从而将gmjagged1
‑
1基因敲除。
[0114]
该材料的gmjagged1
‑
2基因在靶点1位置上缺失序列表序列2第235
‑
238位的核苷酸catg,共缺失4bp,在靶点2位置上,缺失序列表序列2第393位的核苷酸a,共缺失1bp,导致cds起始密码子缺失且第二个外显子移码突变,最终致使gmjagged1
‑
2功能丧失,产生gmjagged1
‑
2基因。
[0115]
三、gmjagged1
‑
1和gmjagged1
‑
2双突变体jag2
‑
7产量相关性状测定
[0116]
将t1代jag2
‑
7自交,收获t2代jag2
‑
7种子。将t2代jag2
‑
7的大豆种子和野生型大豆华春6号(wt),于3月初和6月初种植于广东省广州市华南农业大学试验基地的大豆隔离区内,株距12cm,行距50cm,行长1.0m,设三个生物学重复,每个生物学重复3行。在收获季节分别统计单株粒数和单株粒重。
[0117]
结果见图2,在春季和夏季两个生长季节,与受体材料华春6号相比,jag2
‑
7的单株粒数均显著的增加,分别增加了17.63%和10.59%。与受体材料华春6号相比,jag2
‑
7的单株粒重均显著的增加,分别增加了8.81%和8.67%。
[0118]
以上对本发明进行了详述。对于本领域技术人员来说,在不脱离本发明的宗旨和范围,以及无需进行不必要的实验情况下,可在等同参数、浓度和条件下,在较宽范围内实施本发明。虽然本发明给出了特殊的实施例,应该理解为,可以对本发明作进一步的改进。总之,按本发明的原理,本技术欲包括任何变更、用途或对本发明的改进,包括脱离了本技术中已公开范围,而用本领域已知的常规技术进行的改变。按以下附带的权利要求的范围,可以进行一些基本特征的应用。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1