一种2-甲胺基-4-甲氧基-6-甲基-1,3,5-三嗪的合成方法与流程
一种2
‑
甲胺基
‑4‑
甲氧基
‑6‑
甲基
‑
1,3,5
‑
三嗪的合成方法
技术领域
1.本发明涉及化学合成技术领域,具体为一种2
‑
甲胺基
‑4‑
甲氧基
‑6‑
甲基
‑
1,3,5
‑
三嗪的合成方法。
背景技术:
2.苯磺隆是杜邦公司在二十世纪八十年代初期开发的一种重要的磺酰脲类除草剂,是我国麦田防阔叶杂草的当家品种。具有高效、广谱、低毒、高选择性等特点。苯磺隆是侧链氨基酸合成抑制剂,抑制缬氨酸和异亮氨酸的生物合成,从而阻止植物细胞分裂。目前合成路线多为2
‑
甲胺基
‑4‑
甲氧基
‑6‑
甲基
‑
1,3,5
‑
三嗪与邻甲酸甲酯苯磺酰异氰酸酯缩合而成,而2
‑
甲胺基
‑4‑
甲氧基
‑6‑
甲基
‑
1,3,5
‑
三嗪是合成苯磺隆的关键中间体,关于它的合成有大量的报道,但是生产工艺都存在条件苛刻、危险、原料成本高等问题,因此探索一条经济合理的工艺路线对其工业化生产具有重要意义。2
‑
甲胺基
‑4‑
甲氧基
‑6‑
甲基
‑
1,3,5
‑
三嗪的化学结构式如下:。
3.目前,2
‑
甲胺基
‑4‑
甲氧基
‑6‑
甲基
‑
1,3,5
‑
三嗪报道的合成工艺路线主要如下:美国专利us4933450、us6342600b1公布了以双氰胺钠为起始原料,经过醋酸锌或氯化锌催化醇解、环合和氨解反应得到2
‑
甲胺基
‑4‑
甲氧基
‑6‑
甲基
‑
1,3,5
‑
三嗪。此工艺路线反应主原料昂贵不易得,工艺收率低,产品成本偏高,所以该工艺路线没有市场竞争力。
4.美国专利us4886881a、中国专利cn104387334公布了以双氰胺为起始原料,经过醋酸锌或氯化锌催化醇解、环合和甲基化得到2
‑
甲胺基
‑4‑
甲氧基
‑6‑
甲基
‑
1,3,5
‑
三嗪。此工艺路线虽然原料便宜,但是反应转化率低,产品成本还是偏高,也没有市场竞争力。
5.中国专利cn102295614公布了以乙腈为起始原料,经过氯气氯代、环合、取代和氨解反应得到2
‑
甲胺基
‑4‑
甲氧基
‑6‑
甲基
‑
1,3,5
‑
三嗪。此工艺路线虽然原料易得,但是设备要求高,用到氯气和氯化氢,腐蚀性强,污染严重,也不太适合规模化生产。
6.世界专利wo9811076a1公布了以三聚氯氰为起始原料,经过与甲基氯化镁进行格氏试剂交换、甲醇钠取代和氨解得到2
‑
甲胺基
‑4‑
甲氧基
‑6‑
甲基
‑
1,3,5
‑
三嗪。此工艺路线缺点是甲基氯化镁价格昂贵,反应浓度较稀,产能偏低,需要无水无氧反应条件苛刻,产品成本较高没有市场竞争力。
技术实现要素:
7.为克服了上述问题,本发明公开了一种2
‑
甲胺基
‑4‑
甲氧基
‑6‑
甲基
‑
1,3,5
‑
三嗪的合成方法,工艺过程简单、原料易得、生产成本较低,易于规模化生产。
8.为了达到以上目的,本发明提供如下技术方案:
2
‑
甲胺基
‑4‑
甲氧基
‑6‑
甲基
‑
1,3,5
‑
三嗪的结构式如式i所示:。
9.一种2
‑
甲胺基
‑4‑
甲氧基
‑6‑
甲基
‑
1,3,5
‑
三嗪的合成方法,具体步骤如下:步骤1:将三聚氯氰、化合物iii和溶剂a加入反应容器中,控制温度,缓慢加入缚酸剂,保温搅拌,制得化合物iv;步骤2:将化合物iv加入溶剂b中,控制温度,缓慢加入甲醇钠,保温搅拌,制得化合物v溶液;在化合物v溶液中加入碱,控制温度,保温搅拌,制得化合物vi;步骤3:在化合物vi中加入溶剂c,控制温度,然后滴加40%甲胺溶液,保温搅拌,制得化合物i。
10.所述合成方法的反应式如下:所述合成方法的反应式如下:;其中,r为c1
‑
c12的烷基、苯基、苄基中的一种。
11.进一步地,所述步骤1中三聚氯氰与化合物iii及缚酸剂的质量比为1:1~2:2~3,所述三聚氯氰与溶剂a的质量比为1:4~6;所述步骤1中的控制温度为
‑
20~20℃,优选地,控制温度为
‑
15~0℃;所述步骤1中保温搅拌的时间为1~12小时,优选地,保温搅拌的时间为1~5小时。
12.进一步地,所述步骤1中的溶剂a为丙酮、丁酮、甲基叔丁酮、甲基异丁酮、乙酸乙酯、二氯甲烷、二氯乙烷、四氯化碳、甲苯、二甲苯、苯、乙苯、异丙基苯、氯苯、正己烷、环己烷、十二烷、四氢呋喃、氯仿、乙腈、n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺、n,n
‑
二乙基甲酰胺、1,4
‑
二氧六环、n
‑
甲基吡咯烷酮、二甲基亚砜、环丁砜中的一种。
13.进一步地,所述步骤1中的缚酸剂为钠氢、碳酸钠、碳酸钾、碳酸铯、氢氧化钠、氢氧化钾、甲醇钠、乙醇钠、叔丁醇钠、叔丁醇钾、叔丁醇锂中的一种。
14.进一步地,所述步骤2中化合物iv与甲醇钠的摩尔比为1:2~3,所述化合物iv与溶剂b的质量比为1:4~8;所述步骤2中的化合物iv至化合物v反应的控制温度为0~30℃,优选地,控制温度为0~10℃,保温搅拌的时间为1~12小时,优选地,温搅拌的时间为8~10小时。
15.进一步地,所述步骤2中的溶剂b为甲醇、乙醇、乙二醇、异丙醇、n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺、n,n
‑
二乙基甲酰胺、丁酮、甲基叔丁酮、乙酸乙酯、二氯甲烷、二氯乙烷、四氯化碳、甲苯、二甲苯、苯、乙苯、异丙基苯、氯苯、正己烷、环己烷、十二烷、四氢呋喃、氯仿、乙腈、1,4
‑
二氧六环、n
‑
甲基吡咯烷酮、二甲基亚砜、环丁砜中的一种。
16.进一步地,所述步骤2中化合物iv与碱的摩尔比为1:1~2;所述步骤2中的化合物v至化合物vi反应的控制温度为0~100℃,优选地,控制温度为0~30℃;保温搅拌的时间为1~12小时,优选地,保温搅拌的时间为6~12小时。
17.进一步地,所述步骤2中的碱为碳酸氢钠、碳酸氢钾、碳酸钠、碳酸钾、碳酸铯、氢氧化钠、氢氧化钾、氢氧化钙、氧化钙、磷酸钾、磷酸氢二钾、磷酸钠、磷酸氢二钠、醋酸钠、醋酸钾、甲醇钠、乙醇钠、叔丁醇钠、叔丁醇钾、叔丁醇锂中的一种。
18.进一步地,所述步骤3中化合物vi与甲胺的摩尔比为1:1~2,所述化合物vi与溶剂c的质量比为1:1~3;所述步骤3中的控制温度为0~30℃,优选地,控制温度为20~30℃;所述步骤3中的保温搅拌的时间为1~12小时,优选地,保温搅拌的时间为10~12小时。
19.进一步地,所述步骤3中的溶剂c为甲醇、乙醇、乙二醇、异丙醇、n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺、n,n
‑
二乙基甲酰胺、丁酮、甲基叔丁酮、乙酸乙酯、二氯甲烷、二氯乙烷、四氯化碳、甲苯、二甲苯、苯、乙苯、异丙基苯、氯苯、正己烷、环己烷、十二烷、四氢呋喃、氯仿、乙腈、1,4
‑
二氧六环、n
‑
甲基吡咯烷酮、二甲基亚砜中的一种。
20.与现有技术相比,本发明具有如下优点和有益效果:(1)本发明的反应条件温和,对反应设备没有特殊要求,无危险工艺。
21.(2)本发明原材料易得,工艺操作简单,适合工业规模化生产。
22.(3)本发明采用叠缩工艺,降低了生产成本,产品具有市场竞争力。
附图说明
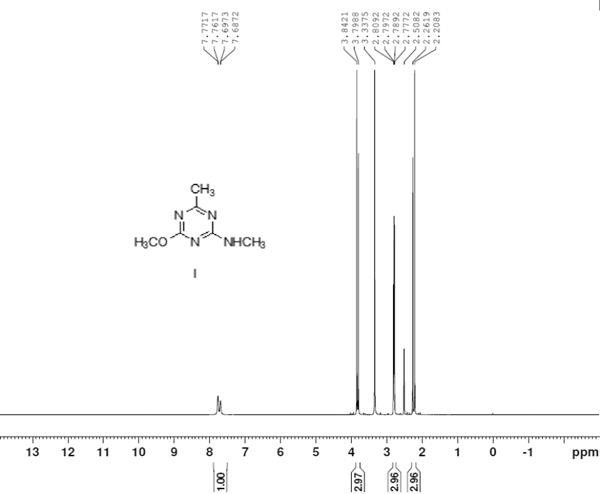
23.图1为本发明实施例1中间体iv的hnmr图谱;图2为本发明实施例3中间体v的hnmr图谱;图3为本发明实施例3中间体vi的hnmr图谱;图4为本发明实施例5化合物i的hnmr图谱。
具体实施方式
24.以下将结合具体实施例对本发明提供的技术方案进行详细说明,应理解下述具体实施方式仅用于说明本发明而不用于限制本发明的范围。
25.实施例1中间体(iv)的制备:氩气保护,在反应瓶中依次加入100g三聚氯氰(ii),500g溶剂a,开搅拌,再加入73.1g丙二酸二甲酯(iii),冰盐降温至
‑
5℃,分批加入61.5g甲醇钠,控制加入温度小于0℃,保温反应1
‑
12小时,hplc检测原料<0.5%,中控合格后,减压回收溶剂,补加200g水,用2n盐酸调ph<3,充分搅拌30分钟,过滤,得黄色固体,50℃鼓风干燥,得中间体(iv)106
‑
144g,纯度≥98%,收率69.8
‑
95%。1hnmr(cdcl3,400mhz):δ3.86(s,6h,och3),δ5.02(s,1h,ch);中间体iv的hnmr图谱如图1所示。以各具体的溶剂a进行实验并调整反应时间,结果如
表1所示。
26.表1 不同溶剂a对中间体iv收率的影响实施例2中间体(iv)的制备:氩气保护,在反应瓶中依次加入100g三聚氯氰(ii),500g四氢呋喃,开搅拌,再加入78g丙二酸二甲酯(iii),冰盐降温至
‑
10℃,缓慢加入缚酸剂,控制加入温度小于0℃,加毕,保温反应1
‑
12小时,hplc检测原料<0.5%,中控合格后,减压回收溶剂,补加200g水,用2n盐酸调ph<3,充分搅拌30分钟,过滤,得黄色固体,50℃鼓风干燥,得中间体(iv)114
‑
150g,纯度≥98%,收率75
‑
99%。以各具体的缚酸剂进行实验并调整当量和反应时间,其中,摩尔比为缚酸剂与三聚氯氰的摩尔比,各结果如表2所示。
27.表2不同缚酸剂对中间体iv收率的影响实施例3中间体(vi)的制备:在反应瓶中依次加入120g中间体(iv),720g溶剂b,开搅拌,冰盐降温至0
‑
5℃,滴加30%甲醇钠溶液163g,滴毕保温1小时,接着缓慢升至室温搅拌1
‑
12小时,hplc中控原料<1.0 %,降温至0℃以下,滴加320ml水,再滴加10% naoh溶液223g,25℃搅拌10小时,用2n稀盐酸调节ph值6
‑
7。hplc中控中间体(v)<0.5%,结束反应,减压回收溶剂,剩余水相用300ml二氯甲烷分两次萃取,合并有机相,40℃减压蒸干得中间体(vi)63
‑
67g,纯度≥95%,收率94
‑
99%。中间体v的hnmr图谱如图2所示,1hnmr(dmso,400mhz):δ3.46(s,6h,och3)δ3.72(s,6h,och3);中间体vi的hnmr图谱如图3所示,1hnmr(dmso,400mhz):δ3.34(s,3h,ch3)δ3.93(s,6h,och3);以各具体的溶剂b进行实验并调整反应时间,各结果如表3所示。
28.表3 不同溶剂b对中间体vi收率的影响
实施例4中间体(vi)的制备:在反应瓶中依次加入120g中间体(iv),720g甲醇,开搅拌,冰盐降温至0
‑
5℃,滴加30%甲醇钠溶液163g,滴毕保温1小时,接着缓慢升至室温搅拌8小时,hplc中控原料<1.0 %,降温至0℃以下,滴加320ml水,再滴加碱,25℃搅拌1
‑
12小时,用2n稀盐酸调节ph值6
‑
7。hplc中控中间体(v)<0.5%,结束反应,减压回收溶剂,剩余水相用300ml二氯甲烷分两次萃取,合并有机相,40℃减压蒸干得中间体(vi)60
‑
66g,纯度≥96%,收率90
‑
99%。以各具体的碱进行实验并调整当量和反应时间,其中,摩尔比为碱与化合物v的摩尔比,各结果如表4所示。
29.表4不同碱对中间体vi收率的影响实施例5 2
‑
甲胺基
‑4‑
甲氧基
‑6‑
甲基
‑
1,3,5
‑
三嗪的制备:在反应瓶中依次加入中间体(vi)60g,180g溶剂c,搅拌,降温至10℃以下,滴加40%甲胺水溶液60g,滴加结束,在20
‑
25℃保温8
‑
12小时,hplc中控中间体(vi)<1%,过滤,50℃鼓风干燥,得 2
‑
甲胺基
‑4‑
甲氧基
‑6‑
甲基
‑
1,3,5
‑
三嗪50
‑
57g,hplc纯度≥98%,收率85
‑
95%。1hnmr(dmso,400mhz):δ2.21~2.26(d,3h,ch3),δ2.78~2.81(m,3h,nch3),δ3.80~3.84(d,3h,och3),δ7.69~7.77 (m,1h,nh)。2
‑
甲胺基
‑4‑
甲氧基
‑6‑
甲基
‑
1,3,5
‑
三嗪的hnmr图谱如图4所示;以各具体的溶剂b进行实验并调整反应时间,各结果如表5所示。
30.表5不同溶剂c对2
‑
甲胺基
‑4‑
甲氧基
‑6‑
甲基
‑
1,3,5
‑
三嗪的收率的影响
实施例6 2
‑
甲胺基
‑4‑
甲氧基
‑6‑
甲基
‑
1,3,5
‑
三嗪的制备:在反应瓶中依次加入中间体(vi)60g,甲醇180g,搅拌,降温至10℃以下,滴加40%甲胺水溶液,滴加结束,在20
‑
25℃保温1
‑
12小时,hplc中控中间体(vi)<1%,过滤,50℃鼓风干燥,得 2
‑
甲胺基
‑4‑
甲氧基
‑6‑
甲基
‑
1,3,5
‑
三嗪50
‑
57g,hplc纯度≥98%,收率90
‑
95%。以各具体的甲胺水的不同摩尔比进行实验调整反应时间,其中,摩尔比为甲胺与化合物vi的摩尔比,各结果如表6所示。
31.表6添加不同量的甲胺对2
‑
甲胺基
‑4‑
甲氧基
‑6‑
甲基
‑
1,3,5
‑
三嗪的收率的影响本发明方案所公开的技术手段不仅限于上述实施方式所公开的技术手段,还包括由以上技术特征任意组合所组成的技术方案。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1