一种天然产物菲式生物碱苷类化合物H4的全合成方法与流程
一种天然产物菲式生物碱苷类化合物h4的全合成方法
技术领域
1.本发明属于有机合成和药物化学领域,具体涉及一种天然产物菲式生物碱苷类化合物h4的全合成方法。
2.背景内容非酒精性脂肪肝(nafld)是肝脏相关发病率和死亡率的最常见原因,全球范围内nafld的患病率估计约为20
‑
30%。nafld是由肝脏脂质堆积引起的,当脂肪含量超过总肝脏重量10%以上,就会出现脂肪肝的迹象。nafld常常与外周胰岛素抵抗、2型糖尿病、肥胖和血脂异常有关。但是,尚无中国、欧盟和美国等国家的食品药品监督管理局批准的药物可以治疗这种疾病。饮食和运动的生活方式干预仍然是非酒精性脂肪肝患者治疗的主流。虽然生活方式干预在实现时是有效的,但是患者的医从性较差。现如今,市场上治疗nafld药物主要有胰岛素增敏剂二甲双胍,降脂药物非诺贝特,抗氧化剂维生素e。虽然这些药物对治疗nafld有一定的效果,但是它们的副作用也是明显的。因此,从丰富的天然药物中寻找高效低毒的治疗nafld药物的研究已成为当前国际医药界的研究热点。
3.传统中药用药历史悠久,疗效确切,蕴藏着丰富的化学多样性信息,是创新药物的重要来源。小花清风藤是传统的保肝中药,其在治疗脂肪肝疾病方面显示出独特的功效。本课题组前期从小花清风藤中分离得到一个结构新颖的菲式生物碱苷类化合物,命名为h4。并对其进行了活性测定,体内外药理实验表明,h4呈剂量依赖性改善非酒精性脂肪肝的病理学症状和生化指标,具有较强的抗非酒精性脂肪肝作用。
4.并且研究显示,由于化合物h4在小花清风藤中含量较低,因此,实践中限制了对其的深入开发与利用,因此研究者试图通过化学合成方法制备化合物h4。本技术的发明人经查阅文献,迄今,尚未见关于化合物h4的全合成方法的报道。
技术实现要素:
5.本发明的目的是针对上述问题,提供一种天然产物菲式生物碱苷类化合物h4的全合成方法,以3
‑
甲氧基
‑4‑
羟基苯乙胺盐酸盐为原料合成化合物h4,原料易得,中间体稳定,反应容易控制,对大量制备化合物h4具有重要的参考和实用价值。
6.本发明是通过以下技术方案实现的:一种天然产物菲式生物碱苷类化合物h4的全合成方法,以3
‑
甲氧基
‑4‑
羟基苯乙胺盐酸盐为原料合成化合物h4,依次包括以下11步反应:酸胺缩合反应、环合反应、硼氢化钠还原反应、boc保护胺基反应、heck反应、脱保护基boc反应、埃施魏勒
‑
克拉克反应、季铵化反应、霍夫曼消除反应、柯尼希斯
‑
克诺尔苷化反应、脱保护基反应。
7.进一步地,所述化合物h4的结构式为:
,其合成路线为:。
8.进一步地,合成化合物h4具体包括以下步骤:(1)合成化合物1酸胺缩合反应:取原料3
‑
甲氧基
‑4‑
羟基苯乙胺盐酸盐溶于无水n,n
‑
二甲基甲酰
胺中,依次加入n,n
‑
二异丙基乙胺、1
‑
乙基
‑
(3
‑
二甲基氨基丙基)碳酰二亚胺盐酸盐、1
‑
羟基苯并三唑、邻溴苯乙酸,搅拌均匀,在氮气保护下,室温反应18
‑
24h,tlc检测反应后,加水淬灭,使用盐酸调ph至中性,然后用有机溶剂萃取,合并有机相,洗涤有机相,干燥,滤除干燥剂,减压挥发有机溶剂,硅胶柱层析,得n
‑
(4
‑
羟基
‑3‑
甲氧基苯乙基)
‑2‑
苯基乙酰胺,即为化合物1;(2)合成化合物2环合反应:取化合物1溶于乙腈,然后加入三氯氧磷,搅拌均匀,在氮气保护下,加热至75
‑
85℃,回流反应6
‑
8h后,加入冰水淬灭,用氢氧化钠溶液调ph至8
‑
9,然后用有机溶剂萃取,合并有机相,洗涤有机相,干燥,滤除干燥剂,减压浓缩,硅胶柱层析,得1
‑
(2
‑
溴苄基)
‑6‑
甲氧基
‑
3,4
‑
二氢异喹啉
‑7‑
醇,即为化合物2;(3)合成化合物3硼氢化钠还原反应:取化合物2溶于甲醇中,然后加入硼氢化钠,置于冰浴中反应2
‑
4h,加水淬灭,调ph至8
‑
9,然后用有机溶剂萃取,合并有机相,洗涤有机相,干燥,滤除干燥剂,减压浓缩,硅胶柱层析,得1
‑
(2
‑
溴苄基)
‑6‑
甲氧基
‑
1,2,3,4
‑
四氢异喹啉
‑7‑
醇,即为化合物3;(4)合成化合物4boc保护胺基反应:取化合物3溶于二氯甲烷中,加入二碳酸二叔丁酯,搅拌均匀,在室温下反应2
‑
4h,加水淬灭,盐酸调滤液ph至中性,然后用有机溶剂萃取,合并有机相,洗涤有机相,干燥,滤除干燥剂,减压浓缩,硅胶柱层析,得1
‑
(2
‑
溴苄基)
‑7‑
羟基
‑6‑
甲氧基
‑
3,4
‑
二氢异喹啉
‑2‑
羧酸叔丁酯,即为化合物4;(5)合成化合物5heck反应:取化合物4溶于无水n,n
‑
二甲基乙酰胺中,依次加入碳酸铯和氯(2
‑
二环己基膦基
‑
2',4',6'
‑
三异丙基
‑
1,1'
‑
联苯基)[2
‑
(2'
‑
氨基
‑
1,1'
‑
联苯)]钯,搅拌均匀,在氮气保护下,在室温中搅拌反应20
‑
40min,然后在100℃下冷凝回流10
‑
14h,tlc检测反应后,加水淬灭,使用盐酸调ph至中性,然后用有机溶剂萃取,合并有机相,洗涤有机相,干燥,滤除干燥剂,旋蒸、冷冻干燥,硅胶柱层析,得1
‑
羟基
‑2‑
甲氧基
‑
6a,7
‑
二氢
‑
4h
‑
二苯并[de,g]喹啉
‑6‑
羧酸叔丁酯,即为化合物5;(6)合成化合物6脱保护基boc反应:取化合物5溶于无水乙醇中,在冰水浴中加入3mol/l的盐酸乙醇溶液,搅拌均匀,在氮气保护下,在0℃反应2
‑
4h,加水淬灭,然后用有机溶剂反向萃取,有机相用水萃取,合并水层,所得水层用氢氧化钠溶液调节ph至8
‑
9,再用有机溶剂萃取,合并有机相,洗涤有机相,干燥,滤除干燥剂,减压浓缩,硅胶柱层析,得2
‑
甲氧基
‑
5,6,6a,7
‑
四氢
‑
4h
‑
二苯并[de,g]喹啉
‑1‑
醇,即为化合物6;(7)合成化合物7埃施魏勒
‑
克拉克反应:取化合物6溶于甲醇中,加入37%(质量百分数)甲醛溶液,然后加入硼氢化钠,置于冰浴中反应4
‑
6h,加水淬灭,盐酸调滤液ph至中性,然后用有机溶剂萃取,合并有机相,洗涤有机相,干燥,滤除干燥剂,减压浓缩,硅胶柱层析,得2
‑
甲氧基
‑6‑
甲基
‑
5,6,6a,7
‑
四氢
‑
4h
‑
二苯并[de,g]喹啉
‑1‑
醇,即为化合物7;(8)合成化合物8
季铵化反应:取化合物7溶于无水丙酮,加入碘甲烷,在氮气保护下,反应12
‑
16 h, tlc检测反应,抽滤得1
‑
羟基
‑2‑
甲氧基
‑
6,6
‑
二甲基
‑
5,6,6a,7
‑
四氢
‑
4h
‑
二苯并[de, g]喹啉
‑6‑
碘化铵,即为化合物8;(9)合成化合物9霍夫曼消除反应:取化合物8溶于水,加入氢氧化钾,在氮气保护下,在70
‑
80℃冷凝回流反应10
‑
14h,加水淬灭,调ph至8
‑
9,然后用有机溶剂萃取,合并有机相,洗涤有机相,干燥,滤除干燥剂,减压浓缩,硅胶柱层析,得1
‑
(2
‑
(二甲基氨基)乙基)
‑3‑
甲氧基
‑4‑
菲酚,即为化合物9;(10)合成化合物10柯尼希斯
‑
克诺尔苷化反应:取化合物9溶于二氯甲烷,依次加入四丁基溴化铵、水、氢氧化钠、2,3,4,6
‑
四乙酰氧基
‑
alpha
‑
d
‑
吡喃葡萄糖溴化物,在氮气保护下,室温反应16
‑
20 h,加水淬灭,然后用有机溶剂萃取,合并有机相,洗涤有机相,干燥,滤除干燥剂,减压浓缩,中性氧化铝柱层析,得1
‑
(2
‑
(二甲基氨基)乙基)
‑3‑
甲氧基
‑4‑
菲酚
‑4‑
o
‑
β
‑
d
‑
乙酰葡萄糖苷,即为化合物10;(11)合成化合物h4脱保护基反应:取化合物10溶于甲苯,加入碘化镁,在氮气保护以及65
‑
70℃下,搅拌反应20
‑
40min,然后缓慢升温至80℃继续反应10
‑
14 h,加水淬灭,然后用有机溶剂萃取,合并有机相,洗涤有机相,干燥,滤除干燥剂,减压浓缩,得粗产物,将粗产物溶于甲醇,加入甲醇钠,在氮气保护及冰浴环境中搅拌反应4
‑
6 h,tlc检测反应后,加水淬灭,然后用有机溶剂萃取,合并有机相,洗涤有机相,干燥,滤除干燥剂,减压浓缩,硅胶柱层析,得1
‑
(2
‑
(二甲基氨基)乙基)
‑
3,4
‑
菲二醇
‑4‑
o
‑
β
‑
d
‑
葡萄糖苷,即为化合物h4。
[0009]
进一步地,步骤(1)中,3
‑
甲氧基
‑4‑
羟基苯乙胺盐酸盐、n,n
‑
二异丙基乙胺、1
‑
乙基
‑
(3
‑
二甲基氨基丙基)碳酰二亚胺盐酸盐、1
‑
羟基苯并三唑、邻溴苯乙酸的摩尔比为1:3.1:1.1:1.1:1.05;步骤(2)中,化合物1与三氯氧磷的摩尔比为1:(3~5.5);步骤(3)中,化合物2与硼氢化钠的摩尔比为1:(1.2~4);步骤(4)中,化合物3与二碳酸二叔丁酯的摩尔比为1:(1~3);步骤(5)中,化合物4、碳酸铯、氯(2
‑
二环己基膦基
‑
2',4',6'
‑
三异丙基
‑
1,1'
‑
联苯基)[2
‑
(2'
‑
氨基
‑
1,1'
‑
联苯)]钯的摩尔比为1:(2~3):0.1;步骤(6)中,化合物5与盐酸乙醇溶液中盐酸的摩尔比为1:10;步骤(7)中,化合物6、甲醛、硼氢化钠的摩尔比为1:4:8;步骤(8)中,化合物7与碘甲烷的摩尔比为1:1.6;步骤(9)中,化合物8与氢氧化钾的摩尔比为1:(10~20);步骤(10)中,化合物9、四丁基溴化铵、氢氧化钠、2,3,4,6
‑
四乙酰氧基
‑
alpha
‑
d
‑
吡喃葡萄糖溴化物的摩尔比为1:1.1:4.9:2;步骤(11)中,化合物10、碘化镁、甲醇钠的摩尔比为1:(4~7):(4~7)。
[0010]
进一步地,步骤(1)、(4)、(5)、(7)中的盐酸质量浓度为1
‑
5%,步骤(2)、(6)中的氢氧化钠的质量浓度为1
‑
3%。
[0011]
进一步地,步骤(3)中调节ph至8
‑
9的方法为:先用1%盐酸调节ph至3
‑
4,后用2%氢
氧化钠溶液调节ph至8
‑
9;步骤(9)中调节ph至8
‑
9的方法为:先用1%盐酸调节ph至4
‑
5,后用2%氢氧化钠调节ph至8
‑
9。
[0012]
进一步地,步骤(1)、(2)、(4)、(6)、(7)、(10)、(11)中用于萃取的有机溶剂为乙酸乙酯,步骤(3)、(5)、(9)中用于萃取的有机溶剂为二氯甲烷,步骤(6)中反向萃取的有机溶剂为乙酸乙酯。
[0013]
进一步地,步骤(1)、(2)、(3)、(5)、(6)、(7)、(9)、(10)、(11)中洗涤有机相的试剂为饱和氯化钠溶液,步骤(4)中洗涤有机相的试剂依次为饱和碳酸氢钠溶液和饱和氯化钠溶液,步骤(1)、(2)、(3)、(4)、(5)、(6)、(7)、(9)、(10)、(11)中干燥过程使用的干燥剂为无水硫酸钠。
[0014]
进一步地,步骤(1)、(2)、(3)、(6)、(7)、(9)、(11)中硅胶柱层析所用洗脱剂为二氯甲烷/甲醇,其中步骤(1)、(7)、(9)中二氯甲烷/甲醇的体积比为40:1,步骤(2)、(3)、(6)中二氯甲烷/甲醇的体积比为30:1,步骤(11)中二氯甲烷/甲醇的体积比为10:1;步骤(4)、(5)中硅胶柱层析所用洗脱剂为石油醚/乙酸乙酯,其中石油醚/乙酸乙酯的体积比分别为6:1、8:1;步骤(10)中,中性氧化铝柱层析所用洗脱剂为二氯甲烷/甲醇,二氯甲烷/甲醇的体积比为200:1。
[0015]
进一步地,步骤(3)、(7)冰浴反应过程中,还包括向反应物中滴加甲醇。
[0016]
与现有技术相比,本发明具备以下有益效果:本发明提供的合成方法,可以用于合成天然产物菲式生物碱苷类化合物h4,且合成路线简单,反应过程稳定易控制,产率较高,克服了天然产物提取分离过程繁琐,产率低的缺点。
附图说明
[0017]
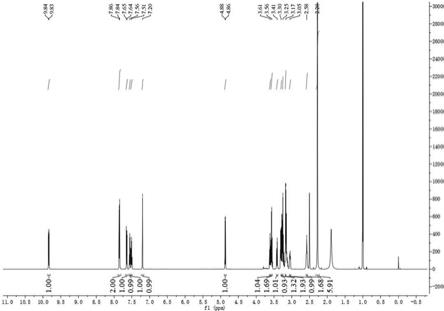
图1为本发明合成的天然产物菲式生物碱苷类化合物h4的1h
‑
nmr图谱;图2为本发明合成的天然产物菲式生物碱苷类化合物h4的
13
c
‑
nmr图谱;图3为本发明中间产物化合物1的1h
‑
nmr图谱;图4为本发明中间产物化合物1的
13
c
‑
nmr图谱;图5为本发明中间产物化合物2的1h
‑
nmr图谱;图6为本发明中间产物化合物2的
13
c
‑
nmr图谱;图7为本发明中间产物化合物3的1h
‑
nmr图谱;图8为本发明中间产物化合物3的
13
c
‑
nmr图谱;图9为本发明中间产物化合物4的1h
‑
nmr图谱;图10为本发明中间产物化合物4的
13
c
‑
nmr图谱;图11为本发明中间产物化合物5的1h
‑
nmr图谱;图12为本发明中间产物化合物5的
13
c
‑
nmr图谱;图13为本发明中间产物化合物6的1h
‑
nmr图谱;图14为本发明中间产物化合物6的
13
c
‑
nmr图谱;图15为本发明中间产物化合物7的1h
‑
nmr图谱;图16为本发明中间产物化合物7的
13
c
‑
nmr图谱;图17为本发明中间产物化合物8的1h
‑
nmr图谱;
(co), 146.6 (c
‑
3), 144.2 (c
‑
4), 134.7 (c
‑1′
), 133.1 (c
‑3′
), 131.6 (c
‑6′
), 130.3 (c
‑
1), 129.1 (c
‑4′
), 127.9 (c
‑5′
), 124.9(c
‑2′
), 121.2 (c
‑
6), 114.2 (c
‑
2), 111.0 (c
‑
5), 55.9 (och3), 44.1 (ch2co), 40.9 (c
‑
α), 35.1 (ch2‑
β)。
[0021]
(2)合成化合物2取化合物1 8.900 g(24.4 mmol)加入到250 ml茄型烧瓶中,溶于40.0 ml乙腈,加入三氯氧磷20.642 g(134.5 mmol),搅拌均匀,在氮气保护下,加热至80℃,回流反应7h。将茄型烧瓶移至冰水浴之中,向反应液缓慢加入20.0 ml冰水,用2%氢氧化钠溶液调节ph至8.5,期间溶液变浑浊,有固体析出。使用乙酸乙酯萃取(30.0 ml
×
3),合并乙酸乙酯。用25.0 ml饱和nacl溶液洗涤乙酸乙酯层,用无水na2so4干燥。减压浓缩得棕色液体,使用硅胶柱层析(二氯甲烷:甲醇=30:1)纯化化合物,得到1
‑
(2
‑
溴苄基)
‑6‑
甲氧基
‑
3,4
‑
二氢异喹啉
‑7‑
醇7.440 g(21.5 mmol),收率为88.1%。该过程如下反应:。
[0022]
化合物2的结构表征数据为:esi
‑
ms m/z:347[m+h]
+
。1h
‑
nmr (600 mhz, cd3od): δ 7.62 (1h, dd, j = 1.2 and 7.8 hz, h
‑3′
), 7.27 (1h, t, j = 7.2 hz, h
‑5′
), 7.20 (1h, d, j = 7.2 hz, h
‑6′
), 7.16 (1h, m, h
‑4′
), 7.02 (1h, s, h
‑
8), 6.88 (1h, s, h
‑
5), 3.92 (3h, s, och3), 3.67 (2h, m, h
‑
3), 2.79 (2h, m, h
‑
α), 1.32 (2h, m, ha
‑
4); 13
c nmr (150 mhz, cd3od) δ: 168.3 (c
‑
1), 151.4 (c
‑
6), 145.1 (c
‑
7), 136.2 (c
‑1′
), 132.7 (c
‑3′
), 130.9 (c
‑6′
), 130.2 (c
‑
8a), 128.5 (c
‑4′
), 127.5 (c
‑5′
), 124.3 (c
‑
4a), 120.7 (c
‑2′
), 113.3 (c
‑
5), 110.2 (c
‑
8), 55.1 (och3), 46.4 (c
‑
α), 45.5 (c
‑
3), 25.0 (c
‑
4)。
[0023]
(3)合成化合物3取化合物2 8.460 g(24.5 mmol)加入到100 ml茄型烧瓶中,溶于30.0 ml甲醇。在恒压滴液漏斗加入20.0 ml甲醇,然后再加入硼氢化钠1.858 g(28.9 mmol)。将反应装置置于冰浴之中,将恒压滴液漏斗中的甲醇缓慢滴加入反应瓶中,反应3 h。向反应液加入20.0 ml水进行淬灭反应,滴加1%盐酸调节ph至3.5,后用2%氢氧化钠溶液调节ph至8.5,反应溶液变浑浊,有固体析出。然后用二氯甲烷萃取(30.0 ml
×
3),合并二氯甲烷。用25.0 ml饱和nacl溶液洗涤二氯甲烷层,用无水na2so4干燥。减压浓缩得淡黄色液体,使用硅胶柱层析(二氯甲烷:甲醇=30:1)纯化化合物,得到1
‑
(2
‑
溴苄基)
‑6‑
甲氧基
‑
1,2,3,4
‑
四氢异喹啉
‑7‑
醇6.740 g(19.4 mmol),收率为79.2%。该过程如下反应:
27.8 (3
×
ch3)。
[0027]
(5)合成化合物5取化合物4 6.180 g(13.8 mmol)加入到250 ml茄型烧瓶中,溶于40 ml无水n,n
‑
二甲基乙酰胺(dma),依次加入cs2co
3 9.000 g(27.6 mmol)和氯(2
‑
二环己基膦基
‑
2',4',6'
‑
三异丙基
‑
1,1'
‑
联苯基)[2
‑
(2'
‑
氨基
‑
1,1'
‑
联苯)]钯(xphos
·
pdg2)1.085 g(1.38 mmol),搅拌均匀,在氮气保护下,在室温中搅拌30min,然后在100℃下冷凝回流12 h,tlc检测反应。
[0028]
使反应液冷却至室温,加入20.0 ml水淬灭反应。缓慢滴加1%盐酸调节ph至中性,加水,然后用二氯甲烷萃取(30.0 ml
×
3),合并二氯甲烷。用25.0 ml饱和nacl溶液洗涤二氯甲烷层,用无水na2so4干燥。旋蒸去除溶剂,再用冷冻干燥去除多余的dma,得到黑色固体。使用硅胶柱层析(石油醚:乙酸乙酯=8:1)纯化化合物,得到1
‑
羟基
‑2‑
甲氧基
‑
6a,7
‑
二氢
‑
4h
‑
二苯并[de, g]喹啉
‑6‑
羧酸叔丁酯 3.200g(8.7mmol),收率为63.0%。该过程如下反应:。
[0029]
化合物5的结构表征数据为:esi
‑
ms m/z:448[m+h]
+
。1h
‑
nmr (600 mhz, cdcl3): δ 8.41 (1h, d, j = 6.0 hz, h
‑
11), 7.32 (1h, t, j = 6.0 hz, h
‑
9), 7.24 (1h, d, j = 6.0 hz, h
‑
8), 7.32 (1h, m, h
‑
10), 6.60 (1h, s, h
‑
3), 6.17 (1h, br s, 1
‑
oh), 4.71 (1h, d, j = 12.0 hz h
‑
6a), 4.40 (1h, s, ha
‑
5), 3.91 (3h, s, och3‑
2), 2.94 (2h, m, c
‑
4), 2.83 (2h, t, j = 12.0 hz, c
‑
7), 2.60(1h, d, j = 18.0 hz, hb
‑
5), 1.49 (9h, s, 3
×
ch3). 13
c
‑
nmr (150 mhz, cdcl3): δ 154.7 (co), 145.6 (c
‑
2), 141.8 (c
‑
1), 136.6 (c
‑
7a), 131.9 (c
‑
11a), 128.5 (c
‑
3a), 128.0 (c
‑
11), 127.2 (2c, c
‑
11b, c
‑
8), 126.6 (c
‑
10), 125.0 (c
‑
9), 120.0 (c
‑
11c), 109.5 (c
‑
3), 79.8 (oc
‑
tbu), 56.2 (och3), 51.6 (c
‑
6a), 38.6 (c
‑
5), 35.4 (c
‑
4), 30.1 (c
‑
7), 28.6 (3
×
ch3)。
[0030]
(6)合成化合物6取化合物5 3.200 g(8.7 mmol)加入到100ml茄型烧瓶中,溶于10.0 ml无水乙醇,在冰水浴中加入3m hcl
·
ch3ch2oh 29.2 ml(hcl,87 mmol),搅拌均匀,在n2保护下,在0℃反应3 h。加入20.0 ml水淬灭反应,然后加入30.0 ml乙酸乙酯反向萃取,乙酸乙酯层用水萃取(30.0 ml
×
3),合并水层。所得水层用2%%氢氧化钠溶液调节ph至8.5,溶液变浑浊,有白色固体析出。水相用乙酸乙酯萃取(30.0 ml
×
3),合并乙酸乙酯。用25.0 ml饱和nacl溶液洗涤乙酸乙酯层,用无水na2so4干燥,减压浓缩得到透明状胶状液体。使用硅胶柱层析(二氯甲烷:甲醇=30:1)纯化化合物,得到2
‑
甲氧基
‑
5,6,6a,7
‑
四氢
‑
4h
‑
二苯并[de, g]喹啉
‑1‑
醇1.82g(6.8mmol),收率为78.2%。该过程如下反应:
ml饱和nacl溶液洗涤乙酸乙酯层,用无水na2so4干燥。减压浓缩得棕色液体,使用硅胶柱层析(二氯甲烷:甲醇=30:1)纯化化合物,得到1
‑
(2
‑
溴苄基)
‑6‑
甲氧基
‑
3,4
‑
二氢异喹啉
‑7‑
醇。
[0044]
(3)合成化合物3取化合物2 5.283 g(15.3 mmol)加入到100 ml茄型烧瓶中,溶于30.0 ml甲醇。在恒压滴液漏斗加入25.0 ml甲醇,然后再加入硼氢化钠2.449 g(38.1 mmol)。将反应装置置于冰浴之中,将恒压滴液漏斗中的甲醇缓慢滴加入反应瓶中,反应2h。向反应液加入20.0 ml水进行淬灭反应,滴加1%盐酸调节ph至3,后用2%氢氧化钠溶液调节ph至8,反应溶液变浑浊,有固体析出。然后用二氯甲烷萃取(30.0 ml
×
3),合并二氯甲烷。用25.0 ml饱和nacl溶液洗涤二氯甲烷层,用无水na2so4干燥。减压浓缩得淡黄色液体,使用硅胶柱层析(二氯甲烷:甲醇=30:1)纯化化合物,得到1
‑
(2
‑
溴苄基)
‑6‑
甲氧基
‑
1,2,3,4
‑
四氢异喹啉
‑7‑
醇。
[0045]
(4)合成化合物4取化合物3 10.461 g(30.0 mmol)加入到100 ml茄型烧瓶中,溶于25.0 ml二氯甲烷,加入二碳酸二叔丁酯6.543 g(30.0 mmol),搅拌均匀,在室温中反应2h。向反应液加入20.0 ml水进行淬灭反应,使用3%盐酸调滤液ph至中性,然后用乙酸乙酯萃取(30.0 ml
×
3),合并乙酸乙酯。有机相依次用饱和nahco3(20 ml)和饱和nacl水溶液(20 ml)洗涤乙酸乙酯层,用无水na2so4干燥。减压浓缩得到透明状胶状液体。使用硅胶柱层析(石油醚:乙酸乙酯=6:1)纯化化合物,得到1
‑
(2
‑
溴苄基)
‑7‑
羟基
‑6‑
甲氧基
‑
3,4
‑
二氢异喹啉
‑2‑
羧酸叔丁酯。
[0046]
(5)合成化合物5取化合物4 5.329 g(11.9 mmol)加入到250 ml茄型烧瓶中,溶于40 ml无水n,n
‑
二甲基乙酰胺(dma),依次加入cs2co
3 9.685 g(29.7 mmol)和氯(2
‑
二环己基膦基
‑
2',4',6'
‑
三异丙基
‑
1,1'
‑
联苯基)[2
‑
(2'
‑
氨基
‑
1,1'
‑
联苯)]钯(xphos
·
pdg2)0.936 g(1.19 mmol),搅拌均匀,在氮气保护下,在室温中搅拌20min,然后在100℃下冷凝回流10h,tlc检测反应。
[0047]
使反应液冷却至室温,加入20.0 ml水淬灭反应。缓慢滴加3%盐酸调节ph至中性,加水,然后用二氯甲烷萃取(30.0 ml
×
3),合并二氯甲烷。用25.0 ml饱和nacl溶液洗涤二氯甲烷层,用无水na2so4干燥。旋蒸去除溶剂,再用冷冻干燥去除多余的dma,得到黑色固体。使用硅胶柱层析(石油醚:乙酸乙酯=8:1)纯化化合物,得到1
‑
羟基
‑2‑
甲氧基
‑
6a,7
‑
二氢
‑
4h
‑
二苯并[de, g]喹啉
‑6‑
羧酸叔丁酯。
[0048]
(6)合成化合物6取化合物5 3.200 g(8.7 mmol)加入到100ml茄型烧瓶中,溶于10.0 ml无水乙醇,在冰水浴中加入3m hcl
·
ch3ch2oh 29.2 ml(hcl,87 mmol),搅拌均匀,在n2保护下,在0℃反应2h。加入20.0 ml水淬灭反应,然后加入30.0 ml乙酸乙酯反向萃取,乙酸乙酯层用水萃取(30.0 ml
×
3),合并水层。所得水层用1%%氢氧化钠溶液调节ph至8,溶液变浑浊,有白色固体析出。水相用乙酸乙酯萃取(30.0 ml
×
3),合并乙酸乙酯。用25.0 ml饱和nacl溶液洗涤乙酸乙酯层,用无水na2so4干燥,减压浓缩得到透明状胶状液体。使用硅胶柱层析(二氯甲烷:甲醇=30:1)纯化化合物,得到2
‑
甲氧基
‑
5,6,6a,7
‑
四氢
‑
4h
‑
二苯并[de, g]喹啉
‑1‑
醇。
[0049]
(7)合成化合物7
取化合物6 2.397 g(9.0 mmol)加入到100 ml茄型烧瓶中,溶于10.0 ml甲醇,加入2.617 ml 37%甲醛溶液(36.0 mmol)。在恒压滴液漏斗加入32.0 ml甲醇,然后再加入硼氢化钠2.729 g(72.0 mmol)。将反应装置于冰浴之中,将恒压滴液漏斗中的甲醇滴加入反应瓶中,反应4h。加入20.0 ml水淬灭反应,使用3%盐酸调滤液ph至中性,然后用乙酸乙酯萃取(30.0 ml
×
3),合并乙酸乙酯。用25.0 ml饱和nacl溶液洗涤乙酸乙酯层,用无水na2so4干燥,减压浓缩得到透明状胶状液体。使用硅胶柱层析(二氯甲烷:甲醇=40:1)纯化化合物,得到2
‑
甲氧基
‑6‑
甲基
‑
5,6,6a,7
‑
四氢
‑
4h
‑
二苯并[de, g]喹啉
‑1‑
醇。
[0050]
(8)合成化合物8取化合物7 1.200 g(2.8 mmol)加入到50 ml茄型烧瓶中,溶于10.0 ml无水丙酮,加入0.637 g碘甲烷(4.5 mmol),在氮气保护下,反应12h,发现大量白色固体析出,tlc检测反应。抽滤,得白色固体粗品1
‑
羟基
‑2‑
甲氧基
‑
6,6
‑
二甲基
‑
5,6,6a,7
‑
四氢
‑
4h
‑
二苯并[de, g]喹啉
‑6‑
碘化铵。
[0051]
(9)合成化合物9取化合物8 1.744 g(6.25 mmol)加入到50 ml茄型烧瓶中,溶于5.0 ml水,加入2.311 g氢氧化钾(62.55 mmol),在氮气保护下,在70℃下冷凝回流下反应10h。加水淬灭,向反应液加1%盐酸调节ph至4,后用2%氢氧化钠调节ph至8,溶液产生浑浊,有固体析出。后用二氯甲烷萃取(30.0 ml
×
3),合并二氯甲烷。用25.0 ml饱和nacl溶液洗涤二氯甲烷层,用无水na2so4干燥,,减压浓缩得到贴壁透明固体。使用硅胶柱层析(二氯甲烷:甲醇=40:1)纯化化合物,得到1
‑
(2
‑
(二甲基氨基)乙基)
‑3‑
甲氧基
‑4‑
菲酚。
[0052]
(10)合成化合物10取化合物9 0.200 g(0.7 mmol)加入到25ml茄型烧瓶中,溶于5.0 ml二氯甲烷,依次加入0.262 g四丁基溴化铵(0.8 mmol),5.0 ml水,0.136g氢氧化钠(3.4 mmol),0.557 g 2,3,4,6
‑
四乙酰氧基
‑
alpha
‑
d
‑
吡喃葡萄糖溴化物(1.4 mmol)。在氮气保护下,室温反应16h。反应液使用乙酸乙酯萃取(15.0 ml
×
3),合并乙酸乙酯。用20.0 ml饱和nacl溶液洗涤乙酸乙酯层,用无水na2so4干燥,减压浓缩得到透明固体。使用中性氧化铝柱层析(二氯甲烷:甲醇=200:1)纯化化合物,得到1
‑
(2
‑
(二甲基氨基)乙基)
‑3‑
甲氧基
‑4‑
菲酚
‑4‑
o
‑
β
‑
d
‑
乙酰葡萄糖苷。
[0053]
(11)合成化合物h4取化合物10 0.412 g(0.22 mmol)加入到25 ml茄型烧瓶中,溶于甲苯,加入0.254 g碘化镁(0.89 mmol),在n2保护下,在65℃搅拌反应20min,然后缓慢升温至80℃继续反应10 h。反应液加10.0 ml水淬灭,然后用乙酸乙酯萃取(30.0 ml
×
3),合并乙酸乙酯,用20.0 ml饱和nacl溶液洗涤乙酸乙酯层,用无水na2so4干燥,减压浓缩得到红棕色干燥粗产物固体。将粗产物转移至25 ml茄型烧瓶中,溶于5.0 ml甲醇,加入0.048 g甲醇钠(0.89 mmol)。在氮气保护下,在冰浴环境中搅拌反应4 h。运用tlc检测反应,合成所得化合物在365 nm有蓝色荧光,且与从植物中提取得到的h4具有相同的rf。反应液加10.0 ml水淬灭,然后使用乙酸乙酯萃取(30.0 ml
×
3),合并乙酸乙酯,用20.0 ml饱和nacl溶液洗涤乙酸乙酯层,用无水na2so4干燥,减压浓缩得到白色固体。使用硅胶柱层析(二氯甲烷:甲醇=10:1)纯化化合物,得到1
‑
(2
‑
(二甲基氨基)乙基)
‑
3,4
‑
菲二醇
‑4‑
o
‑
β
‑
d
‑
葡萄糖苷。
[0054]
实施例3
2,3,4,6
‑
四乙酰氧基
‑
alpha
‑
d
‑
吡喃葡萄糖溴化物(1.4 mmol)。在氮气保护下,室温反应20h。反应液使用乙酸乙酯萃取(15.0 ml
×
3),合并乙酸乙酯。用20.0 ml饱和nacl溶液洗涤乙酸乙酯层,用无水na2so4干燥,减压浓缩得到透明固体。使用中性氧化铝柱层析(二氯甲烷:甲醇=200:1)纯化化合物,得到1
‑
(2
‑
(二甲基氨基)乙基)
‑3‑
甲氧基
‑4‑
菲酚
‑4‑
o
‑
β
‑
d
‑
乙酰葡萄糖苷。
[0065]
(11)合成化合物h4取化合物10 0.243 g(0.13 mmol)加入到25 ml茄型烧瓶中,溶于甲苯,加入0.268g碘化镁(0.94 mmol),在n2保护下,在70℃搅拌反应40min,然后缓慢升温至80℃继续反应14 h。反应液加10.0 ml水淬灭,然后用乙酸乙酯萃取(30.0 ml
×
3),合并乙酸乙酯,用20.0 ml饱和nacl溶液洗涤乙酸乙酯层,用无水na2so4干燥,减压浓缩得到红棕色干燥粗产物固体。将粗产物转移至25 ml茄型烧瓶中,溶于5.0 ml甲醇,加入0.050 g甲醇钠(0.94 mmol)。在氮气保护下,在冰浴环境中搅拌反应6 h。运用tlc检测反应,合成所得化合物在365 nm有蓝色荧光,且与从植物中提取得到的h4具有相同的rf。反应液加10.0 ml水淬灭,然后使用乙酸乙酯萃取(30.0 ml
×
3),合并乙酸乙酯,用20.0 ml饱和nacl溶液洗涤乙酸乙酯层,用无水na2so4干燥,减压浓缩得到白色固体。使用硅胶柱层析(二氯甲烷:甲醇=10:1)纯化化合物,得到1
‑
(2
‑
(二甲基氨基)乙基)
‑
3,4
‑
菲二醇
‑4‑
o
‑
β
‑
d
‑
葡萄糖苷。
[0066]
最后应说明的是,以上所述仅为本发明的优选实施例而已,并不用于限制本发明,尽管参照前述实施例对本发明进行了详细的说明,对于本领域的技术人员来说,其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1