一种紫外光刺激响应可控润湿性交联分子刷及其制备方法与流程
1.本发明涉及刺激响应材料技术领域,具体涉及一种紫外光刺激响应可控润湿性交联分子刷及其制备方法。
背景技术:
2.随着社会经济不断发展,人们对于材料表面性能的要求也日新月异。表面改性可以赋予材料表面各种特性,包括化学惰性、粘附性、生物相容性、亲水性、疏水性等。利用接枝聚合物分子刷对材料表面进行改性具有高效、简便的特点,并已经成为调控界面物理和化学性质重要的改性办法之一。
3.刺激响应材料是一种能够对外界条件做出响应,并呈现相应性能的新型智能材料。将刺激响应单元引入聚合物分子刷中,响应前后材料表面性能会发生显著变化。可控交联/解交联聚合物分子刷即是刺激响应材料的一种,目前对可控交联分子刷的研究报道还较少,且刺激方式多为ph、温度刺激等为主,如公开号为cn101560062a的专利公开刺激响应性聚合物刷的制备方法,该专利制得的聚合物刷具有温度刺激响应性,但这些刺激方式不仅可控性较差,且耗费大量热能或者溶剂使其不符合可持续发展的理念。
技术实现要素:
4.本发明所要解决的技术问题在于提供一种具有紫外光刺激响应的交联分子刷及其制备方法。
5.本发明通过以下技术手段实现解决上述技术问题:
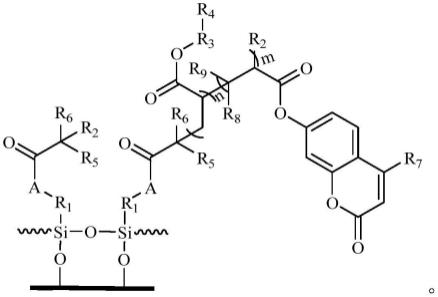
6.一种紫外光刺激响应可控润湿性交联分子刷,其结构式如下:
[0007][0008]
其中r1选自c
1-12
烷基、c
1-12
烷氧基、c
2-12
链烯基;r2选自br、cl;r3选自c
1-12
烷基、c
1-12
烷氧基、c
2-12
链烯基;r4选自全氟丁基、全氟己基、全氟癸基;r5和r6相同或相异,并独立选自c
1-2
烷基;r7选自氢原子、c
1-12
烷基、c
1-12
烷氧基、c
2-12
链烯基;r8和r9相同或相异,并独立选自氢原子、c
1-12
烷基、c
1-12
烷氧基、c
2-12
链烯基、卤素原子、氰基、c
6-10
芳基、c
6-10
芳氧基、c
6-10
芳烷氧基、c
8-12
芳基链烯基、c
3-8
环烷基、羧基、羧基c
1-12
烷基酯基、羧基聚(c
1-4
)烷撑二醇醚酯
基、c
2-7
羧基烷氧基、c
1-12
烷基酯基、c
2-7
羧基烷氧基聚(c
1-4
)烷撑二醇醚酯基;
[0009]
a选自于
[0010]
m为20-45之间的整数;
[0011]
n为15-30之间的整数;
[0012]
为聚多巴胺改性固态基体。
[0013]
有益效果:本发明中的紫外光刺激响应可控润湿性交联分子刷能够利用香豆素基团的交联和解交联,赋予表面可控的亲疏水性。
[0014]
本发明中的紫外光刺激响应可控润湿性交联分子刷经过365nm紫外光照之后,香豆素基团交联,赋予材料表面好的抗蛋白质黏附的特性。
[0015]
优选地,所述r1为正丙基;所述的r2为br;所述的r3为乙基;所述的r4为全氟丁基;所述r5和r6相同为甲基;所述的r7为甲基;所述r8和r9相同为氢原子;a为
[0016]
优选地,所述紫外光刺激响应可控润湿性交联分子刷的表面交联和解交联过程为365nm紫外光照10-120min,在254nm紫外光照1-10min。
[0017]
制备上述紫外光刺激响应可控润湿性交联分子刷的方法,包括以下步骤:
[0018]
(1)制备聚多巴胺改性固态基体:将固态基体清洗后,干燥,将乙醇溶液、有机溶剂1加载到固态基体表面,然后加入多巴胺盐酸盐,搅拌反应结束后得到涂覆有聚多巴胺涂层的固体基体;
[0019]
(2)聚多巴胺改性固态基体接枝物质c’:将聚多巴胺改性固态基体放入容器内,加入有机溶剂2,60℃搅拌后加入物质c’,继续在60℃下搅拌反应,得到c’接枝的聚多巴胺改性固态基体;
[0020]
所述物质c’为通式iv的化合物:
[0021]
其中r1、r2、r5、r6和a的定义和紫外光刺激响应可控润湿性交联分子刷结构式中相同;
[0022]
(3)紫外光刺激响应可控润湿性交联分子刷的制备:将称取物质g’、物质f’、催化剂a和显色剂b加入施伦克瓶中,然后向其中加入有机溶剂3、去离子水,向溶液中通氮气除去溶解氧;将c’接枝的聚多巴胺改性固态基体加入另一个施伦克瓶中,抽真空通氮气将体系中的空气置换成氮气氛;然后将上述除氧操作后的施伦克瓶中混合液用注射器转移到充满氮气氛的施伦克瓶中,在氮气保护下,反应后,洗去未接枝的单体,然后氮气氛干燥,得到产品;
[0023]
所述物质f’为通式vii的化合物:其中r7,r8,r9的定义和紫外光刺激响应可控润湿性交联分子刷结构式中相同;
[0024]
所述物质g’为通式viii的化合物:其中r3、r4的定义和紫外光刺激响应可控润湿性交联分子刷结构式中相同。
[0025]
有益效果:本发明后以聚多巴胺(pda)作为二次官能化平台,利用si-atrp法将香豆素衍生物单体与含氟丙烯酸酯单体共聚到材料表面,通过香豆素基团在365nm和254nm紫外光下发生交联和解交联反应,来实现材料表面的亲疏水可控转换,并且聚合物分子刷处于交联状态的表面具有较强的抗蛋白质粘附性能。
[0026]
优选地,所述步骤(1)中有机溶剂1为碱性物质,优选为氨水。
[0027]
优选地,所述固态基体为玻璃片,采用去离子水和有机溶剂4清洗固态基体。
[0028]
优选地,所述有机溶剂4能够溶剂反应物,优选为丙酮。
[0029]
优选地,所述步骤(1)中有机溶剂2能溶解物质c’,优选为40%乙醇溶液。
[0030]
优选地,所述步骤(1)中将涂覆有聚多巴胺涂层的固体基体取出后,分别用无水乙醇和去离子水反复冲洗,以除去材料表面非特异性沉积的聚多巴胺粒子,然后在氮气氛中吹干备用,剩余的反应溶液通过旋蒸以除去无水乙醇和去离子水。
[0031]
优选地,所述步骤(2)中物质c’由物质物质a’与物质b’反应获得;
[0032]
所述物质a’为通式ii的化合物:
[0033][0034]
所述物质b’为通式iii的化合物:
[0035]
其中r2,r5和r6和定义和紫外光刺激响应可控润湿性交联分子刷结构式中相同;r
11
为br或cl,优选为br。
[0036]
优选地,所述物质c’的制备方法具体包括以下步骤:将有机溶剂5、物质a’、有机溶剂6加入充满氮气的烧瓶中,冰水浴搅拌混合;再将物质b’缓慢滴入其中,反应1小时后,升至室温反应24小时后结束;过滤除去白色沉淀后旋蒸除去有机溶剂,然后减压蒸馏除去剩余溶剂和未反应原料,得到棕色液体,即为产物c’。
[0037]
优选地,所述有机溶剂5能够溶解物质a’与物质b’,优选为甲苯;优选地,所述有机溶剂6能够溶解物质a’与物质b’,优选为三乙胺。
[0038]
优选地,所述物质a’和b’的摩尔比为1:(0.5-2.0),优选为1:(1.2-1.8),所述物质a’与有机溶剂5的摩尔比为1:(0.5-2.0),优选为1:(1.2-1.8)。
[0039]
优选地,所述步骤(3)中催化剂a为n,n,n
′
,n
″
,n
″‑
五甲基二亚乙基三胺,催化剂a物质的量为0.3mmol。
[0040]
优选地,所述步骤(3)中显色剂b能够用来指示反应是否完成,优选为溴化亚铜。
[0041]
优选地,所述步骤(3)中采用去离子水和有机溶剂9洗去未接枝的单体。
[0042]
优选地,所述步骤(3)中物质f’由物质d’与物质e’反应获得;
[0043]
所述物质d’为通式v的化合物:
[0044]
其中r7的定义和紫外光刺激响应可控润湿性交联分子刷结构式中相同,r
10
为羟基、氨基、巯基,优选为羟基;
[0045]
所述物质e’为通式vi的化合物:
[0046]
其中r8、r9的定义和紫外光刺激响应可控润湿性交联分子刷结构式中相同。
[0047]
优选地,所述物质f’的制备方法具体包括以下步骤:向三口烧瓶中加入物质d’和三乙胺,然后加入有机溶剂7,冰水浴冷却至0℃,然后搅拌30分钟,使其充分溶解;然后将物质e’溶于有机溶剂8,缓慢滴加到体系中,搅拌1小时后,25℃继续反应2小时结束;饱和食盐水洗三次静置,有机相经干燥剂a干燥,旋蒸除去溶剂,提纯后,得到白色固体,即为物质f’。
[0048]
优选地,所述有机溶剂7能够溶解物质d’,优选为n,n-二甲基甲酰胺;所述有机溶剂8能够溶解物质e’,优选为乙酸乙酯。
[0049]
优选地,所述干燥剂a用来干燥有机相,优选为无水硫酸镁。
[0050]
优选地,所述步骤(3)中物质g’与f’的摩尔比为1:(0-10),优选为1:(0-4)。
[0051]
优选地,所述步骤(3)中物质f’经柱色谱法提纯,优选洗脱剂比例为pe:ea=6:1。
[0052]
本发明的优点在于:本发明中的紫外光刺激响应可控润湿性交联分子刷能够利用香豆素基团的交联和解交联,赋予表面可控的亲疏水性。
[0053]
本发明中的紫外光刺激响应可控润湿性交联分子刷经过365nm紫外光照之后,香豆素基团交联,赋予材料表面好的抗蛋白质黏附的特性。
[0054]
本发明后以聚多巴胺(pda)作为二次官能化平台,利用表面原子转移自由基聚合(si-atrp)法将香豆素衍生物单体与含氟丙烯酸酯单体共聚到材料表面,通过香豆素基团在365nm和254nm紫外光下发生交联和解交联反应,来实现材料表面的亲疏水可控转换,并且聚合物分子刷处于交联状态的表面具有较强的抗蛋白质粘附性能。
附图说明
[0055]
图1为本发明实施例1中聚多巴胺涂覆表面的反应流程图;
[0056]
图2为本发明实施例2中brmpa的1h nmr图;
[0057]
图3为本发明实施例4中amycm的1h nmr图;
[0058]
图4为本发明实施例5制备的cum-f-g表面的xps图谱;
[0059]
图5为本发明实施例6制备的cum-f-g表面的xps图谱;
[0060]
图6为本发明实施例7制备的cum-f-g表面的xps图谱;
[0061]
图7为本发明实施例8制备的cum-f-g表面的xps图谱;
[0062]
图8为本发明实施例5所制备的表面cum8-f2-g光响应可控润湿性循环测试图;
[0063]
图9为本发明实施例5、实施例6、实施例7、实施例8制备的表面的抗蛋白质粘附性图。
具体实施方式
[0064]
为使本发明实施例的目的、技术方案和优点更加清楚,下面将结合本发明实施例,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0065]
下述实施例中所用的试验材料和试剂等,如无特殊说明,均可从商业途径获得。
[0066]
实施例中未注明具体技术或条件者,均可以按照本领域内的文献所描述的技术或条件或者按照产品说明书进行。
[0067]
其中氨丙基三甲氧基硅烷(ar):天津希恩思奥普德科技有限公司;2-溴异丁酰溴(ar):上海毕得医药科技有限公司;三乙胺(tea,ar):天津希恩思奥普德科技有限公司;7-羟基-4甲基香豆素(hmcm,ar):北京伊诺凯科技有限公司;丙烯酰氯(ar):天津希恩思奥普德科技有限公司;多巴胺
·
盐酸盐(pda
·
hcl,ar):天津希恩思奥普德科技有限公司;2-(全氟丁基)乙基丙烯酸酯(pfbea,ar):北京迈瑞达科技有限公司;n,n,n
′
,n",n"-五甲基二亚乙基三胺(pmdeta,ar):上海毕得医药科技有限公司;无水硫酸镁(mgso4,ar):北京化工厂;氯化钠(ar):北京化工厂甲苯(ar):天津市大茂化学试剂厂;无水乙醇(ar):天津市大茂化学试剂厂;乙腈(ar):天津市光复科技发展有限公司;乙酸乙酯(ea,ar):天津市大茂化学试剂厂;石油醚(pe,ar):天津市大茂化学试剂厂。
[0068]
实施例1
[0069]
聚多巴胺改性固态基体的制备
[0070]
利用玻璃刀将实验室用载玻片切成8mm
×
8mm的小片,用去离子水和丙酮反复清洗,氮气氛中干燥备用。
[0071]
将上述处理好的玻璃片放入100ml三口烧瓶中,然后向其中加入65ml 30%乙醇溶液,移液枪取0.8ml氨水加入体系。室温下搅拌半小时使其充分混合,然后称取0.6g的da
·
hcl加入反应体系中,在空气氧化条件下,室温搅拌反应24小时,结束反应,反应流程如图1所示。将涂覆有聚多巴胺涂层的玻璃片取出,分别用无水乙醇和去离子水反复冲洗,以除去材料表面非特异性沉积的聚多巴胺粒子,然后在氮气氛中吹干备用,剩余的反应溶液通过旋蒸以除去无水乙醇和去离子水,得到聚多巴胺涂覆表面pda-glass。
[0072]
实施例2
[0073]
si-atrp引发剂brmpa(物质c’)的合成
[0074]
将50ml无水甲苯,0.90g(5mmol)aps,0.81g(8mmol)无水tea加入充满氮气的烧瓶中,冰水浴搅拌30分钟使其混合均匀。再将1.84g(8mmol)bibb缓慢滴入其中,反应1小时后,
升至室温反应24小时后结束。过滤除去白色沉淀后旋蒸除甲苯,然后减压蒸馏除去剩余溶剂和未反应原料,得到棕色液体,即为brmpa。
[0075][0076]
si-atrp引发剂brmpa的核磁谱图参见图2,其核磁数据如下:1h nmr(400mhz,chloroform-d)δ6.89(s,1h),3.58(d,j=5.7hz,9h),3.27(td,j=7.1,5.9hz,2h),1.95(s,6h),1.66(dddd,j=9.4,5.3,2.5,1.4hz,2h),0.70
–
0.64(m,2h)。
[0077]
实施例3
[0078]
pda-glass表面接枝brmpa
[0079]
将制得的pda-glass放入100ml的烧瓶,随后加入25ml 40%乙醇溶液,60℃下搅拌三十分钟,然后称取0.04g实施例2中制得的brmpa,快速加入反应体系,继续在60℃下搅拌三小时,将pda-glass取出,得到表面brmpa接枝的玻璃片,即br-glass。
[0080][0081]
实施例4
[0082]
7-丙烯酸酯基-4-甲基香豆素(amycm,物质f’)的合成
[0083]
向100ml三口圆底烧瓶中加入0.90g的hmcm(物质d’)和0.51g tea,然后加入4ml dmf,冰水浴冷却至0℃,然后搅拌30分钟,使其充分溶解。然后将0.46g丙烯酰氯(物质e’)溶于15ml ea,缓慢滴加到体系中,搅拌1小时后,25℃继续反应2小时结束。饱和食盐水洗三次静置,有机相经无水硫酸镁干燥,旋蒸除去ea,柱色谱(pe:ea=6:1)提纯,得到白色固体,即为amycm。
[0084][0085]
香豆素衍生物单体amycm的核磁谱图参见图3,其核磁数据如下:1h nmr(400mhz,chloroform-d)δ7.63(d,j=8.7hz,1h),7.20
–
7.10(m,2h),6.66(dd,j=17.3,1.2hz,1h),6.35(dd,j=17.3,10.6hz,1h),6.28(q,j=1.3hz,1h),6.10(dd,j=10.4,1.1hz,1h),2.45(d,j=1.3hz,3h)。
[0086]
实施例5
[0087]
紫外光刺激响应可控润湿性交联分子刷的制备
[0088]
称取0.13g 2-(全氟丁基)乙基丙烯酸酯(pfbea)以及0.37g实施例4中的amycm、五
甲基二乙烯三胺(pmdeta)0.05g(0.3mmol)以及cubr 0.0143g(0.1mmol)加入100ml施伦克瓶中,然后向其中加入22.5ml丙酮,2.5ml去离子水,向溶液中通氮气三十分钟除去溶解氧。将上述atrp引发剂接枝后的材料加入另一个施伦克瓶中,抽真空通氮气操作三次以将体系中的空气置换成氮气氛。然后将上述除氧操作后的施伦克瓶中混合液用注射器转移到充满氮气氛的施伦克瓶中,在氮气保护下,50℃下反应6小时,再用去离子水和丙酮洗去未接枝的单体等,然后氮气氛干燥,得到紫外光刺激响应可控润湿性交联分子刷cum8-f2-g。
[0089][0090]
cum8-f2-g表面的xps图谱如图4所示,cum8-f2-g表面未经过紫外光照射时的水接触角为80
°
,该表面经过365nm紫外光照2h后,香豆素基团交联,cum8-f2-g表面的水接触角上升到107.9
°
。然后再经过254nm紫外光照射10min后,香豆素基团解交联,cum8-f2-g表面的水接触角下降到79.1
°
。
[0091]
实施例6
[0092]
重复实施例5的步骤,制备紫外光刺激响应可控润湿性交联分子刷cum10-f0-g。不同之处在于添加的amycm为0.46g,pfbea为0g。
[0093]
cum10-f0-g表面的xps图谱如图5所示,cum10-f0-g表面未经紫外光照射时的水接触角为73.6
°
,该表面经过365nm紫外光照2h后,香豆素基团交联,cum10-f0-g表面的水接触角上升到102.3
°
。然后再经过254nm紫外光照射10min后,香豆素基团解交联,cum10-f0-g表面的水接触角下降到73.5
°
。
[0094]
实施例7
[0095]
重复实施例5的步骤,制备可控润湿表面材料cum5-f5-g。不同之处在于添加的amycm为0.23g,pfbea为0.32g。
[0096]
cum5-f5-g表面的xps图谱如图6所示,cum5-f5-g表面未经过紫外光照射时的水接触角为87.2
°
,该表面经过365nm紫外光照2h后,香豆素基团交联,cum5-f5-g表面的水接触角上升到114.0
°
。然后再经过254nm紫外光照射10min后,香豆素基团解交联,cum5-f5-g表面的水接触角下降到88.3
°
。
[0097]
实施例8
[0098]
重复实施例5的步骤,制备可控润湿表面材料cum2-f8-g。不同之处在于添加的amycm为0.10g,pfbea为0.52g。
[0099]
cum2-f8-g表面的xps图谱如图7所示,cum2-f8-g表面未经过紫外光照射时的水接
触角为91.0
°
,该表面经过365nm紫外光照2h后,香豆素基团交联,cum2-f8-g表面的水接触角上升到128.0
°
。然后再经过254nm紫外光照射10min后,香豆素基团解交联,cum2-f8-g表面的水接触角下降到92.6
°
。
[0100]
实施例9
[0101]
测定实施例5所制得的表面cum8-f2-g的水接触角。同时利用水接触角测定仪对材料表面紫外光响应可控润湿性进行表征,将cum8-f2-g表面一半用锡箔纸包裹,置于365nm紫外光照射2小时,然后分别测定照射区域和未照射区域的水接触角。然后将cum8-f2-g表面置于254nm光源下照射10分钟,并测定其水接触角。重复该操作三次,以验证可控润湿性的可重复性,结果如图8所示。重复光照三次后,同种状态下的水接触角没有明显差异,说明可控润湿性的可重复性良好。
[0102]
实施例10
[0103]
将实施例5,实施例6,实施例7,实施例8所得到的表面(紫外光刺激前后)浸没在2ml磷酸盐缓冲液(pbs溶液)中2小时,然后再浸入1ml牛血清蛋白标准液(bsa溶液)(4.5mg
·
ml-1
)中,在37℃恒温烘箱中培养2小时后取出。用pbs溶液反复冲洗三次后,将样品置于1wt%十二烷基硫酸钠溶液(sds溶液)中,超声20分钟以使粘附的蛋白质完全脱落,sds溶液待测试。使用bicinchonininc acid(bca)法测定解吸附溶液中的蛋白质含量。将上述溶有bsa的sds溶液稀释10倍后,利用总蛋白质定量测试盒配制工作液,得到待测试样品。测定所有样品在562nm处的吸光度,表征抗蛋白质粘附性。
[0104]
结果如图9所示。从图中可以看到,当材料表面在365nm光照120min后,其表面抗蛋白质吸附能力相较未交联前增强,这是因为365nm紫外光照后,材料表面香豆素衍生物发生环加成反应,分子刷间交联程度增加,限制了蛋白质在表面的粘附。
[0105]
本发明也可以直接采用聚多巴胺粒子接枝brmpa,然后继续制备产物后涂覆于固态基体表面。
[0106]
以上实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的精神和范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1