定向淋巴的前药
或-c2炔基c(o)r
3-基团;
21.r3是自消除基团;
22.x’是o、s、n(r4)或n(h)s(o)2;
23.r4是h或c
1-c4烷基;或者
24.其药学上可接受的盐。
25.在另一方面,本发明提供式(v)表示的式(i)的化合物:
[0026][0027]
其中r1、r2和-x-如对式(i)所定义;
[0028]
r5和r6独立地选自氢和c
1-c4烷基;且
[0029]
r3是自消除基团;或者
[0030]
其药学上可接受的盐。
[0031]
在另一方面,本发明提供一种治疗或预防疾病或障碍的方法,在所说的疾病或障碍中增加的睾酮水平是有益的,该方法包括对有此需要的受试者施用治疗有效量的式(v)的化合物。
[0032]
在另一方面,本发明提供式(v)的化合物在制备用于治疗或预防疾病或障碍的药剂中的用途,在所说的疾病或障碍中增加的睾酮水平是有益的。
[0033]
在另一方面,本发明提供式(v)的化合物,其用于治疗或预防疾病或障碍,在该疾病或障碍中增加的睾酮水平是有益的。
[0034]
在另一方面,本发明提供一种促进药用物质的淋巴转运和系统释放的方法,该方法包括使式(vi)的前药残基或其药学上可接受的盐与药用物质缀合:
[0035][0036]
其中
[0037]
r1和r2独立地表示h或c
2-c
28
脂肪酸的残基;
[0038]-x-选自-o-、-nh-和-s-;
[0039]
当-l-是-x
’‑
时,-y-表示任选被取代的-c
1-c2烷基c(o)r
3-基团或-c2烯基c(o)r
3-或-c2炔基c(o)r
3-基团;
[0040]
r3是自消除基团;以及
[0041]
表示连接基与药物活性剂缀合的点。
[0042]
对于本领域技术人员而言,结合所附的实施例和权利要求书阅读下文的详细描述时,本发明的这些和其它方面将变得更加显见。
附图说明
[0043]
图1:十二指肠内输注睾酮、十一烷酸睾酮(tu)、化合物21、化合物12、化合物13/14、化合物15、化合物17和化合物19后在麻醉的肠系膜淋巴管插套管的雌性sd大鼠中总的睾酮相关衍生物的蓄积淋巴转运(所施用剂量的%)与时间的关系的图解表示。
[0044]
图2:对有意识的颈动脉插套管的雌性sd大鼠经口管饲睾酮、十一烷酸睾酮(tu)、化合物12、化合物15、化合物16、化合物17、化合物18、化合物20和化合物21后剂量归一化睾酮血浆浓度的图解表示。
[0045]
图3:对有意识的颈动脉插套管的雌性sd大鼠经口管饲睾酮、十一烷酸睾酮(tu)、化合物12、化合物13、化合物13/14、化合物18和化合物19后剂量归一化睾酮血浆浓度的图解表示。
[0046]
图4:十二指肠内输注ser盐酸盐(ser.hcl)和化合物3后在麻醉的肠系膜淋巴管插套管的雄性sd大鼠中总的舍曲林(ser)相关衍生物的蓄积淋巴转运(所施用剂量的%)与时间的关系的图解表示。
[0047]
图5:经口管饲ser盐酸盐(ser.hcl)、化合物1、化合物2和化合物3后剂量归一化舍曲林(ser)血浆浓度的图解表示。
[0048]
图6:十二指肠内输注bup、化合物6和化合物7后在麻醉的肠系膜淋巴管插套管的雄性sd大鼠中总的丁丙诺啡(bup)相关衍生物的蓄积淋巴转运(所施用剂量的%)与时间的关系的图解表示。
[0049]
图7:经口管饲bup、化合物5、化合物6和化合物7后剂量归一化丁丙诺啡(bup)血浆浓度的图解表示。
[0050]
图8:十二指肠内输注mpa、化合物10和化合物11后在麻醉的肠系膜淋巴管插套管的雄性sd大鼠中总的麦考酚酸(mpa)相关衍生物的蓄积淋巴转运(所施用剂量的%)与时间的关系的图解表示。
[0051]
图9:与猪胰脂肪酶一起体外温育过程中化合物12、化合物13/14、化合物15、化合物16、化合物17、化合物20和化合物21的单酸甘油酯形式的稳定性特征的图解表示。
[0052]
图10:mpa从1,3-二棕榈酰基甘油麦考酚酸酯(mpa-tg)、化合物10和化合物11中释放的图解表示。
具体实施方式
[0053]
当前药策略用于药物开发领域以改善药物动力学特性时,通常预期前药通过非特异性降解或酶介导的生物转化复原为母体化合物,然后表现出生物活性。本发明公开了基于修饰的甘油酯的化合物,它们能够促进药用物质的淋巴转运并且改善所述化合物复原为活性药用物质。
[0054]
膳食脂质,例如甘油三酯类应用独特的代谢途经以获得进入淋巴(并且最终进入体循环),其完全不同于其它营养物(例如蛋白质和碳水化合物)的代谢途经。在摄入后,膳食甘油三酯类被体腔脂肪酶水解,每个甘油三酯分子释放一个单酸甘油酯和两个脂肪酸。该单酸甘油酯和两个脂肪酸随后被吸收入肠细胞,在那里它们被重新酯化成甘油三酯类。
[0055]
再合成的甘油三酯类被组装成肠脂蛋白(主要是乳糜微粒)且由此形成的乳糜微粒从肠细胞胞吐出来,且随后获得优先进入肠淋巴管。在淋巴管内,乳糜微粒形式的脂质通
过一系列毛细血管、结节和输送管排出,最终在左锁骨下静脉和颈内静脉的连接处排空进入体循环。在进入血液循环后,乳糜微粒中的甘油三酯类优先和有效地被具有高脂蛋白脂肪酶表达的组织(例如脂肪组织、肝和潜在的某些类型的肿瘤组织)吸收。
[0056]
预期脂质模拟化合物与天然甘油三酯具有类似的行为并且被转运至淋巴系统和通过淋巴系统,然后到达体循环。按照这种方式,母体药用物质的药物动力学和药效学特性可以被操纵以增强进入淋巴和淋巴组织,由此通过避免首过代谢提高口服生物利用度(和潜在地肠流出)。脂质模拟化合物还可以促进药物靶向至淋巴、淋巴结和淋巴组织内的部位和高脂质利用和脂蛋白脂酶表达的部位,例如脂肪组织、肝和一些肿瘤。
[0057]
经由体循环转运后易于转化成母体药物的脂质化前药降低了胃肠(gi)道中的游离药物浓度,这可以在减少胃肠刺激、掩蔽味道、促进药物在肠胆汁盐微团中增溶方面(归因于与内源性单酸甘油酯类的类似性)和提高被动膜渗透性方面(通过增加亲脂性)提供益处。脂质化前药还促进在包含单独的脂质或脂质与表面活性剂和/或助溶剂的混合物的脂质载体中溶解,并且这样做使得能以比母体药物可能可行的剂量更大的剂量以药物溶液施用。
[0058]
本发明的发明人令人惊奇地发现,可以修饰将药用物质连接至甘油酯单元的药物-甘油酯缀合物的部分,以改善药物-甘油酯缀合物在gi道中的稳定性,促进转运至肠淋巴,且最终促进药用物质从药用物质-甘油酯前药中释放。因此,通过改变将药用物质连接至甘油酯单元的“连接基”,对于所得到的化合物可以实现最佳的药物动力学特性。
[0059]
本发明人已经发现,即使当连接基是短链连接基(即,如先前报道的琥珀酸),在药用物质与甘油酯单元之间的连接基上结合自消除基团也导致改善的药物系统释放和系统暴露。自消除基团的结合增强了药用物质在体循环中的释放。虽然自消除基团可能会减少淋巴的药物转运,但首次发现,即使在淋巴转运减少的情况下,系统性药物暴露得到增强,这可能是由于体循环中药物释放的增强所致。进一步发现,自消除基团与甘油酯单元之间的连接基上碳原子的甲基取代可以用于增加在胃肠道中的稳定性,增加淋巴转运并且仍然在体循环中保持良好的转化。
[0060]
在本说明书中,应用了本领域技术人员众所周知的许多术语。尽管如此,为了清楚目的,将定义许多术语。
[0061]
在本说明书中,除非另有定义,否则术语“任选被取代的”是指基团可以进一步被一个或多个基团取代,也可以不被这些基团取代,所述基团选自羟基、烷基、烷氧基、烷氧基羰基、烯基、烯氧基、炔基、炔氧基、氨基、氨基酰基、硫代、芳基烷基、芳基烷氧基、芳基、芳氧基、酰基氨基、羧基、氰基、卤素、硝基、磺基、膦酰基、磷酰基氨基、膦基、杂芳基、杂芳氧基、杂环基、杂环氧基、三卤代甲基、五氟乙基、三氟甲氧基、二氟甲氧基、三氟甲硫基、三氟乙烯基、一烷基氨基和二烷基氨基、一(取代的烷基)氨基和二(取代的烷基)氨基、一芳基氨基和二芳基氨基、一杂芳基氨基和二杂芳基氨基、杂环基一和二杂环基、氨基和具有选自烷基、芳基、杂芳基和杂环基的不同取代基的不对称二取代的胺类。
[0062]
如本技术中所用,单独使用或在复合措词中使用的术语“烷基”表示直链或支链烷基。前缀例如“c
2-c
20”用于表示烷基内的碳原子数(在这种情况中为2-20个)。直链和支链烷基的实例包括甲基、乙基、正丙基、异丙基、正丁基、仲丁基、叔丁基、正戊基、己基、庚基、5-甲基庚基、5-甲基己基、辛基、壬基、癸基、十一基、十二烷基和二十二烷基(c
22
)。
[0063]
如本技术中所用,单独使用或在复合措词中使用的术语“烯基”表示包含至少一个碳-碳双键的直链或支链烃残基,包括上文所定义的烯烃类单不饱和烷基、二不饱和烷基或多不饱和烷基。优选地,烯基是直链烯基。前缀例如“c
2-c
20”用于表示烯基内的碳原子数(在这种情况中为2-20个)。烯基的实例包括乙烯基、烯丙基、1-甲基乙烯基、丁烯基、异丁烯基、3-甲基-2-丁烯基、1-戊烯基、1-己烯基、3-己烯基、1-庚烯基、3-庚烯基、1-辛烯基、1-壬烯基、2-壬烯基、3-壬烯基、1-癸烯基、3-癸烯基、1,3-丁二烯基、1,4-戊二烯基、1,3-己二烯基、1,4-己二烯基和5-二十二烯基(c
22
)。
[0064]
如本技术中所用,单独使用或在复合措词中使用的术语“炔基”表示包含至少一个碳-碳三键的直链或支链烃残基。优选地,炔基是直链炔基。前缀例如“c
2-c
20”用于表示烯基内的碳原子数(在这种情况中为2-20个)。
[0065]
如本技术中所用,单独使用或在复合措词中使用的术语例如“杂环”或“杂环基”表示饱和、部分不饱和或完全不饱和单环、双环或稠合多环环系,其包含至少一个选自氮、硫和氧的杂原子。前缀例如“c
5-c
8”用于表示该基团环状部分内的碳原子数(在这种情况中为5-8个)。适合的杂环取代基的实例包括,但不限于吡咯、呋喃、苯并呋喃、苯并噻唑、咪唑、苯并咪唑、咪唑啉、吡唑、吡唑啉、三唑、唑、唑啉、异唑、异唑啉、呋咱、二唑、哌啶、吡啶、嘧啶、哒嗪和吡嗪,它们各自可以进一步被1-3个取代基取代。
[0066]
如本技术中所用,术语例如“芳基”或“芳族环状基团”表示任意单核或多核的、缀合的或稠合的芳族烃基环系的残基。前缀例如“c
5-c
8”用于表示芳基环状部分内的碳原子数(在这种情况中为5-8个)。芳基的实例包括苯基(单核)、萘基(稠合的多核)、联苯基(缀合的多核)和四氢萘基(稠合的多核)。
[0067]
如本技术中所用,术语“连接基”表示对于如本技术中所述的式(i)的化合物而言从“x”到“l”的化合物部分,其使药用物质连接至甘油酯单元。
[0068]
如本技术中所用,术语“自消除基团”定义一种化学部分,其与所述的连接基形成易断裂的键,并与药用物质形成稳定的键,其中与药用物质的键在连接基裂解时变得不稳定。自消除基团的实例包括,但不限于乙缩醛自消除基团、羧基乙缩醛自消除基团、羧基(甲基乙缩醛)自消除基团、对羟基苄基羰基自消除基团、翻转酯自消除基团和三甲基锁自消除基团。许多其它适合的自消除基团在本领域中是已知的,例如c.a.blencowe等人,polym.chem.2011,2,773-790和f.kratz等人在chemmedchem.2008,3(1),20-53中所述。
[0069]
如本技术中所用,术语“药用物质”表示任意药物活性剂或成像(造影)剂,其将得益于通过肠淋巴系统转运,例如以避免首过代谢或用于在淋巴系统内靶向递送。
[0070]
适合的药物活性剂的实例包括,但不限于睾酮、麦考酚酸(mpa)、丁丙诺啡、雌激素类(雌激素)、阿片类例如吗啡、四氢大麻酚(thc)、大麻二酚、美托洛尔、雷洛昔芬、阿法沙龙、他汀类例如阿托伐他汀、喷他佐辛、普萘洛尔、l-dopa、利多卡因、氯丙嗪、舍曲林、阿米替林、去甲替林、喷他佐辛,三硝酸甘油酯、氧烯洛尔、拉贝洛尔、沙丁胺醇、环硫雄醇、美法仑、洛伐他汀、非甾类抗炎药(nsaids,例如阿司匹林、布洛芬、萘普生)、cox-2抑制剂(例如塞来昔布)、皮质类固醇抗炎药(例如泼尼松龙、泼尼松、地塞米松)、抗疟药(例如羟氯喹)、亚硝基脲类、甲氨蝶呤、更生霉素、蒽环类(例如柔红霉素)、丝裂霉素c、博来霉素、光辉霉素、作用于抑免蛋白的药物(例如环孢素、他克莫司、西罗莫司)、柳氮磺吡啶、来氟米特、麦考酚酸酯、阿片样物质、芬戈莫德、多球壳菌素、苯丁酸氮芥、多柔比星、奈拉滨、可的松、地
塞米松、泼尼松、普拉曲沙、长春碱、硼替佐米、奈拉滨、盐酸柔红霉素、氯法拉滨、阿糖胞苷、达沙替尼、甲磺酸伊马替尼、盐酸普纳替尼、硫酸长春新碱、盐酸苯达莫司汀、磷酸氟达拉滨、波舒替尼、尼洛替尼、美琥他辛、卡培他滨、紫杉醇、吉西他滨、氟维司群、他莫昔芬、拉帕替尼、托瑞米芬、伊沙匹隆、艾立布林、阿苯达唑、伊维菌素、乙胺嗪、阿苯达唑、多西环素、氯生太尔、马拉韦罗、恩夫韦肽、脱氧胸腺嘧啶核苷、齐多夫定、司他夫定、去羟肌苷、扎西他滨、阿巴卡韦、拉米夫定、恩曲他滨、替诺福韦、地拉韦啶、利匹韦林、拉替拉韦、艾维雷韦、洛匹那韦、茚地那韦、奈非那韦、氨普那韦、利托那韦、阿昔洛韦和药物活性肽类。
[0071]
适合的成像剂的实例包括,但不限于,荧光团,例如用于荧光显微镜检查术的或用于体内成像的其中荧光团在红外范围内具有发射光谱的alexa fluor系列的光学成像探针;可以用于正电子发射断层摄影术(pet)的γ-发射体,例如氟脱氧葡萄糖或螯合剂,以便螯合磁共振成像探针,例如钆或铁。
[0072]
为避免任何疑问,涉及“相当于直链c
20
烷基基团的长度”是指20个单独键合的碳原子理论上跨越的长度。
[0073]
在本发明的一些优选的实施方案中,以及关于通式(i)或(ii),如下定义中的一个或多个适用:
[0074]
a)r1和r2独立地表示h或c
2-c
28
脂肪酸的残基。
[0075]
b)r1表示h,且r2表示c
2-c
28
脂肪酸的残基。
[0076]
c)r2表示h,且r1表示c
2-c
28
脂肪酸的残基。
[0077]
d)r1和r2各自表示棕榈酸。
[0078]
e)-x-是-o-。
[0079]
f)-x-是-nh-。
[0080]
g)-x-是-s-。
[0081]
h)-l-是-oc(o)-。
[0082]
i)当-l-是-oc(o)-时,-y-表示任选被取代的-c
1-c
20
烷基c(o)och
2-、-c
2-c
20
烯基c(o)och
2-或-c
2-c
20
炔基c(o)och
2-基团;其中烷基、烯基或炔基基团上的碳原子的一个或多个可以被nh、s、o、c
5-c8芳族或脂族环状基团或者c
5-c8芳族或脂族杂环基团替代,条件是所述烷基、烯基或炔基基团不超过相当于直链c
20
烷基基团的长度。
[0083]
j)-y-表示-c
1-c
20
烷基c(o)och
2-、-c
2-c
20
烯基c(o)och
2-或-c
2-c
20
炔基c(o)och
2-基团,其任选被烷基取代。
[0084]
k)-y-表示-c
1-c
20
烷基c(o)och
2-、-c
2-c
20
烯基c(o)och
2-或-c
2-c
20
炔基c(o)och
2-基团,其任选被甲基取代。
[0085]
l)-l-是-x
’‑
。
[0086]
m)-y-表示任选被取代的-c
1-c2烷基c(o)r
3-基团或-c2烯基c(o)r
3-或-c2炔基c(o)r
3-基团。
[0087]
n)-y-表示任选被烷基取代的-c
1-c2烷基c(o)r
3-基团。
[0088]
o)-y-表示任选被甲基取代的-c
1-c2烷基c(o)r
3-基团。
[0089]
p)r3是自消除基团,其选自乙缩醛、羧基乙缩醛、羧基(甲基乙缩醛)、三甲基锁、对羟基苄基羰基或翻转酯自消除基团。
[0090]
n)x'是o。
[0091]
o)x'是s。
[0092]
p)x'是n(r4)。
[0093]
q)x'是n(h)s(o)2。
[0094]
r)r4是h。
[0095]
s)r4是c
1-c4烷基。
[0096]
t)r4是甲基。
[0097]
在一个实施方案中,l是x’,且-y-表示任选被取代的-c1烷基c(o)r
3-。
[0098]
因此,在另一个实施方案中,本发明提供表示为式(ii)的式(i)的化合物:
[0099][0100]
其中
[0101]
r1、r2和-x-如对式(i)所定义;
[0102]
r3是自消除基团;
[0103]
表示药用物质的残基;
[0104]-l-是-x
’‑
;
[0105]
x’是o、s、n(r4)或n(h)s(o)2;
[0106]
r4是h或c
1-c4烷基;
[0107]
r5选自氢和c
1-c4烷基;或者
[0108]
其药学上可接受的盐。
[0109]
在另一个实施方案中,l是x’,且-y-表示任选被取代的-c2烷基c(o)r
3-。
[0110]
因此,在另一个实施方案中,本发明提供表示为式(iii)的式(i)的化合物:
[0111][0112]
其中
[0113]
r1、r2和-x-如对式(i)所定义;
[0114]
r3是自消除基团;
[0115]
表示药用物质的残基;
[0116]-l-是-x
’‑
;
[0117]
x’是o、s、n(r4)或n(h)s(o)2;
[0118]
r4是h或c
1-c4烷基;
[0119]
r5和r6单独地选自氢和c
1-c4烷基;或者
[0120]
其药学上可接受的盐。
[0121]
在另一个实施方案中,式(iii)的化合物选自表1中所列出的那些化合物。
[0122]
表1.式(iii)的化合物:
[0123][0124][0125]1casi=羧基乙缩醛自消除基团;2cmsi=羧基(甲基乙缩醛)自消除基团;3tml=三甲基锁自消除基团;4fsi-5=通过丧失5-碳内酯释放药用物质a的翻转酯自消除基团。
[0126]
在另一个实施方案中,l是
–
oc(o)-,且-y-表示任选被取代的-c
1-c
20
烷基c(o)och
2-、-c
2-c
20
烯基c(o)och
2-或-c
2-c
20
炔基c(o)och
2-基团;其中烷基、烯基或炔基基团上的碳原子的一个或多个可以被nh、s、o、c
5-c8芳族或脂族环状基团或者c
5-c8芳族或脂族杂环基团替代,条件是所述烷基、烯基或炔基基团不超过相当于直链c
20
烷基基团的长度。
[0127]
因此,在另一个实施方案中,本发明提供表示为式(iv)的式(i)的化合物:
[0128][0129]
其中
[0130]
r1、r2和-x-如对式(i)所定义;
[0131]
表示药用物质的残基;
[0132]
r5和r6单独地选自氢和c
1-c4烷基;
[0133]
r7是氢或c
1-c4烷基;且
[0134]
n是0至18;或者
[0135]
其药学上可接受的盐。
[0136]
在另一个实施方案中,式(iv)的化合物选自表2中所列出的那些化合物。
[0137]
表2.式(iv)的化合物:
[0138][0139][0140][0141]
在一个实施方案中,所述的药用物质是睾酮或其衍生物或类似物。睾酮替代疗法(trt)常用于具有性腺功能减退症(特征在于异常低血清睾酮水平的障碍)的患者以便将其血清睾酮水平恢复至正常范围,且由此缓解性腺功能减退症的许多症状,例如情绪紊乱、性功能障碍等。
[0142]
因此,在一个实施方案中,本发明提供表示为式(v)的式(i)的化合物:
[0143][0144]
其中r1、r2和-x-如对式(i)所定义;
[0145]
r5和r6单独地选自氢和c
1-c4烷基;且
[0146]
r3是自消除基团;或者
[0147]
其药学上可接受的盐。
[0148]
在另一个实施方案中,式(v)的化合物选自表3中所列出的那些化合物。
[0149]
表3.式(v)的化合物:
[0150]
[0151][0152]1asi=乙缩醛自消除基团;2tml=三甲基锁自消除基团;3phb=对羟基苄基羰基自消除基团;4fsi-4=通过丧失4-碳内酯释放药用物质a的翻转酯自消除基团;5fsi-5=通过丧失5-碳内酯释放药用物质a的翻转酯自消除基团;6cmsi=羧基(甲基乙缩醛)自消除基团。
[0153]
在另一个实施方案中,本发明提供治疗或预防疾病或障碍的方法,在该疾病或障碍中增加的睾酮水平是有益的,该方法包括对有此需要的受试者施用治疗有效量的根据式(v)的化合物。
[0154]
在还一个实施方案中,本发明提供根据式(v)的化合物在制备用于治疗或预防疾病或障碍的药剂中的用途,在该疾病或障碍中增加的睾酮水平是有益的。
[0155]
在另一个实施方案中,本发明提供式(v)的化合物,其用于治疗或预防疾病或障碍,在该疾病或障碍中增加的睾酮水平是有益的。
[0156]
增加的睾酮水平可能是有益的疾病和障碍包括,但不限于性腺功能减退症、因骨髓衰竭导致的贫血、因肾衰竭导致的贫血、慢性呼吸衰竭、慢性心力衰竭、类固醇依赖性自身免疫疾病、艾滋病消耗、遗传性血管水肿或荨麻疹、末期乳腺癌或绝经。
[0157]
在另一个实施方案中,本发明提供一种促进药用物质的淋巴转运和系统释放的方法,该方法包括使式(vi)的前药残基或其药学上可接受的盐缀合至所述的药用化合物:
[0158][0159]
其中
[0160]
r1和r2独立地表示h或c
2-c
28
脂肪酸的残基;
[0161]-x-选自-o-、-nh-和-s-;
[0162]-y-表示任选被取代的-c
1-c2烷基c(o)r
3-基团或-c2烯基c(o)r
3-或-c2炔基c(o)r
3-基团;
[0163]
r3是自消除基团;且
[0164]
表示连接基与药物活性剂缀合的点。
[0165]
在一个实施方案中,选择如上文所定义的化合物的包含“y”和“r
3”的连接基,以有利于药用物质稳定地转运至肠淋巴。在另一个实施方案中,选择y和r3,以有利于药用物质在淋巴、淋巴细胞、淋巴组织、具有高脂肪酶活性的组织例如脂肪组织、一些癌症、肝脏中或在体循环中释放。在另一个实施方案中,对y和r3进行选择,以有利于药用物质转运至肠淋巴并且有利于药用物质在淋巴、淋巴细胞、淋巴组织、具有高脂肪酶活性的组织例如脂肪组织、一些癌症、肝脏中或在体循环中释放。
[0166]
本发明的化合物可用于稳定地将药用物质转运至肠淋巴并且在淋巴、淋巴细胞、淋巴组织、具有高脂肪酶活性的组织例如脂肪组织、一些癌症、肝脏中或在体循环中释放药用物质。本发明的化合物特别可用于转运和释放得益于避免首过代谢的药用物质,例如,表现出大于50%首过代谢的化合物。在一个实施方案中,预期所述药用物质表现出大于60%的首过代谢。在另一个实施方案中,所述的药用物质表现出大于70%的首过代谢。在另一个实施方案中,所述的药用物质表现出大于80%的首过代谢。在另一个实施方案中,所述的药用物质表现出大于90%的首过代谢。
[0167]
可以得益于稳定地转运至肠淋巴并且在淋巴、淋巴细胞、淋巴组织、具有高脂肪酶活性的组织例如脂肪组织、一些癌症、肝脏中或在体循环中释放的药用物质包括,但不限于睾酮、麦考酚酸、雌激素类(雌激素)、吗啡、四氢大麻酚、大麻二酚、美托洛尔、雷洛昔芬、阿法沙龙、他汀类药物例如阿托伐他汀、喷他佐辛、普萘洛尔、左旋多巴(l-dopa)、丁丙诺啡、咪达唑仑、利多卡因、氯丙嗪、阿米替林、去甲替林、喷他佐辛、硝酸异山梨酯、三硝酸甘油酯、氧烯洛尔、拉贝洛尔、维拉帕米、沙丁胺醇、环硫雄醇、美法仑、洛伐他汀和药物活性肽类。
[0168]
本发明的化合物还可用于所述药用物质在淋巴系统内的靶向释放,例如在淋巴、淋巴细胞和淋巴组织以及具有高脂肪酶活性的组织例如脂肪组织、一些癌症或肝中。
[0169]
可以得益于在淋巴系统或脂肪组织内靶向释放的药用物质包括,但不限于非甾类抗炎药(nsaids,例如阿司匹林、布洛芬、萘普生)、cox-2抑制剂(例如塞来昔布)、皮质类固醇抗炎药(例如泼尼松龙、地塞米松)、抗疟药(例如羟氯喹)、环磷酰胺、ppar激动剂(例如贝特类)、亚硝基脲类、铂、甲氨蝶呤、硫唑嘌呤、巯嘌呤、氟尿嘧啶、放线菌素d、蒽环类、丝裂霉素c、博来霉素、光辉霉素、作用于抑免蛋白的药物(例如环孢素、他克莫司、西罗莫司)、柳氮磺吡啶、来氟米特、麦考酚酸酯、阿片样物质、芬戈莫德、多球壳菌素、苯丁酸氮芥、多柔比星、奈拉滨、可的松、地塞米松、泼尼松、普拉曲沙、长春碱、硼替佐米、噻替派、奈拉滨、盐酸柔红霉素、氯法拉滨、阿糖胞苷、达沙替尼、甲磺酸伊马替尼、盐酸普纳替尼、硫酸长春新碱、盐酸苯达莫司汀、磷酸氟达拉滨、波舒替尼、尼洛替尼、美琥他辛(omacetaxine,mepesuccinate)、阿那曲唑、卡培他滨、来曲唑、紫杉醇、吉西他滨、氟维司群、他莫昔芬、拉帕替尼、托瑞米芬、伊沙匹隆、艾立布林、阿苯达唑、伊维菌素、乙胺嗪、多西环素、氯生太尔、马拉韦罗、恩夫韦肽、脱氧胸腺嘧啶核苷、齐多夫定、司他夫定、去羟肌苷、扎西他滨、阿巴卡韦、拉米夫定、恩曲他滨、替诺福韦、奈韦拉平、地拉韦啶、依非韦伦、利匹韦林、拉替拉韦、艾维雷韦、洛匹那韦、茚地那韦、奈非那韦、氨普那韦、利托那韦、阿昔洛韦和免疫抑制剂例如麦考酚酸、环孢菌素、他克莫司和西罗莫司。
[0170]
作为一般性的策略,本发明的化合物可以通过如下路径之一合成:
[0171][0172]
方案1.通式(ii)的化合物的合成
[0173]
可以使可轻易得自相应丙二酸的二酰氯i与甘油二酯ii在吡啶的存在下反应,得到酸-甘油三酯(酸-tg)iii(参见方案1)。
[0174][0175]
方案2.通式(iii)的化合物的合成
[0176]
在酸酐iv可得到的情况中,可以通过在吡啶的存在下用甘油二酯ii开环生成酸-tg iii(方案2)。当酸酐iv的r5和r6相同时,该方法实施最佳,但当r5和r6彼此不同时,该方法将得到酸-tg iii的区域异构体混合物。因此,其它方法,例如方案3中所概括的方法应当用于这种情况中。
[0177][0178]
方案3.式(iii)的化合物的合成,其中r5=me,r6=h。
[0179]
为了在r5=me且r6=h的具体实例中得到单一区域异构体形式的酸-tg iii,已知
的羧酸v(lienard,b.m.r.等人,org.biomol.chem.2008,6,(13),2282-2292)可以用作起点(参见方案3)。在标准条件下酸v与1,3-dg ii偶联产生tbdps保护的甘油三酯vi,可以将其用tbaf和acoh处理,得到醇vii。然后可以使用两步氧化方法(pcc,然后kmno4)经由中间体醛viii将醇vii转化成期望的酸-tg iii。
[0180][0181]
方案4.通式(iii)化合物的合成,其中r3是乙缩醛自消除(asi)基团,且x’=o。
[0182]
为了合成在药用物质与烷基间隔基之间包含乙缩醛自消除(asi)基团的化合物,必须使带有醇的母体分子官能化并且活化,然后与酸-甘油三酯iii缀合,如上述方案4中所概括的。在乙酐与乙酸的混合物中用dmso处理醇,导致形成(甲硫基)甲基(mtm)醚ix。采用磺酰氯活化mtm醚ix形成推定的亚砜类物质,可以使该亚砜类物质与酸-甘油三酯iii的羧酸酯反应,得到目标化合物x。
[0183][0184]
方案5.式(iii)化合物的合成,其中r3是羧基乙缩醛(casi)或羧基(甲基乙缩醛)(cmsi)自消除基团,且x’是o或n(r4)。
[0185]
在药用物质包含醇、酚或胺(伯或仲)官能团的情况中,可以采用修饰形式的乙缩醛自消除基团,其中包括另外的羧基基团。母体药物与氯甲酸氯烷基酯反应,得到碳酸氯烷基酯或氨基甲酸氯烷基酯xi(参见方案5)。然后通过在回流的甲苯中用衍生自酸-tg iii的
羧酸酯处理来置换卤化物离去基得到目标化合物xii。
[0186][0187]
方案6.式(iii)化合物的合成,其中r3是三甲基-锁(tml)自消除基团,且x’=o、nr4或s(o)2nh。
[0188]
为了合成在药用物质与烷基间隔基之间包含三甲基锁(tml)自消除基团(levine,m.n.;raines,r.t.chem.sci.2012,3,2412-2420)以有利于母体分子的系统释放的前药,必须用tml部分使酸-甘油三酯iii官能化,然后与药用物质缀合,如上述方案6中所概括的。在标准条件下酸-tg iii与tml苯酚xiii偶联,得到甘油三酯xiv,可以在酸性条件下(10-樟脑磺酸)使其脱保护,得到醇xv。依次地首先将醇xv氧化成醛xvi,然后氧化成酸xvii,接着与包含醇、胺或磺酰胺的药用物质在标准条件下偶联,可以得到目标化合物xviii。
[0189][0190]
方案7.式(iii)化合物的合成,其中r3是对羟基苄基羰基(phb)自消除基团,且x’=o、s或nr4。
[0191]
为了合成包含对羟基苄基(phb)羰基自消除基团的化合物,首先将对羟基苄醇(xix)的伯羟基基团保护为甲硅烷基醚,并且使游离酚羟基基团与酸-tg iii偶联,得到phb甘油三酯xxi(参见方案7)。在除去硅保护基后,可以通过用氯甲酸对硝基苯基(pnp)酯处理来活化伯醇xxii,得到碳酸pnp酯xxiii。然后通过与药用物质(a
–
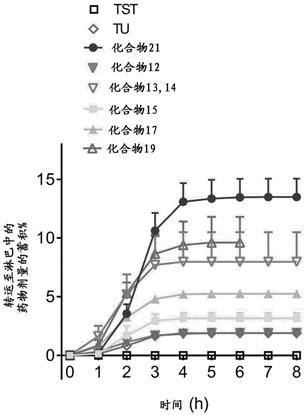
x’h)在碱性条件下反应实现pnp基团的置换,得到期望的化合物xxiv。
[0192][0193]
方案8.式(iii)化合物的合成,其中r3是翻转酯自消除(fsi)基团,且x’=o、s或nr4。
[0194]
设计翻转酯自消除(fsi)基团以通过环化机制,导致4-碳(fsi-4)或5-碳(fsi-5)内酯丧失,释放游离药用物质。可以通过使药用物质(a
–
x’h)与4-溴丁酸(m=1)或5-溴戊酸(m=2)(xxv)偶联来合成fsi前药,得到溴化物xxvi(参见方案8)。采用衍生自酸-tg iii的羧酸酯置换溴化物xxvi生成期望的目标化合物xxvii中的酯键。
[0195][0196]
方案9.式(v)化合物的合成
[0197]
在药用物质包含羧酸的情况中,必须将酸-tg iii转化成相应的酰氯,然后用三烷(r7=h)或三聚乙醛(r7=me)在zrcl4或zncl2的存在下处理,得到氯烷基酯xxviii(参见方案9)。然后可以通过与衍生自母体药物的羧酸酯反应来实现卤化物的置换,得到目标化合物xxix。
[0198]
如果本发明的化合物需要纯化,则可以使用多种技术例如重结晶和色谱技术,包括高效液相色谱法(hplc)和正向或反相硅胶色谱法。所述化合物可以用核磁共振(nmr)质谱法和/或其它适合的方法进行表征。
[0199]
将被理解,本发明的化合物可以以一种或多种立体异构体形式存在(例如非对映异构体)。本发明在其范围内包括所有这些分离(例如对映体分离)或组合(包括外消旋混合物和非对映异构体混合物)的立体异构体形式。
[0200]
因此本发明还涉及基本上纯的立体异构体形式的化合物,例如大于约90%de,例如约95%至97%de或大于99%de,以及混合物,包括其外消旋混合物。这类非对映异构体可以通过不对称合成,例如采用手性中间体来制备,或可以通过常规方法拆分混合物,例如色谱法或应用拆分试剂。
[0201]
如果所述化合物包含一个或多个可被质子化或脱质子化的官能团(例如在生理ph下),则该化合物可以作为药学上可接受的盐制备和/或分离。将被理解,所述化合物在指定ph下可以为两性离子形式。本文所用的表述“药学上可接受的盐”是指所指定的化合物的盐,其中该盐适合于作为药物施用。这类盐可以通过分别使酸或碱与胺或羧酸基团反应形成。
[0202]
药学上可接受的酸加成盐可以由无机酸和有机酸制备。无机酸的实例包括盐酸、氢溴酸、硫酸、硝酸、磷酸等。有机酸的实例包括乙酸、丙酸、乙醇酸、丙酮酸、草酸、苹果酸、丙二酸、琥珀酸、马来酸、富马酸、酒石酸、柠檬酸、苯甲酸、肉桂酸、扁桃酸、甲磺酸、乙磺酸、对甲苯磺酸、水杨酸等。
[0203]
药学上可接受的碱加成盐可以由无机碱和有机碱制备。衍生自无机碱的相应平衡离子包括钠、钾、锂、铵、钙和镁盐。有机碱包括伯胺、仲胺和叔胺类、取代的胺类包括天然存在的取代的胺类和环胺类,包括异丙基胺、三甲胺、二乙胺、三乙胺、三丙胺、乙醇胺、2-二甲
氨基乙醇、氨丁三醇、赖氨酸、精氨酸、组氨酸、咖啡因、普鲁卡因、海巴明(hydrabamine)、胆碱、甜菜碱、乙二胺、葡糖胺、n-烷基葡糖胺、可可碱、嘌呤、哌嗪、哌啶和n-乙基哌啶。
[0204]
酸/碱加成盐比相应的游离酸/碱形式倾向于更溶于水性溶剂。
[0205]
本发明的化合物可以为晶型或为溶剂合物(例如水合物),并且预期两者均在本发明的范围内。术语“溶剂合物”是溶质和溶剂形成的可变化学计算量的复合物。这类溶剂不应当干扰溶质的生物活性。作为实例,溶剂可以为水、乙醇或乙酸。溶剂化方法是本领域中公知的。
[0206]
本发明化合物的施用途经预期包括经口和经肠施用。因此,该活性化合物可以用惰性稀释剂或可同化的可食用载体配制,或可以将其包封在硬壳胶囊或软壳胶囊内,或可以将其压制成片剂,或可以将其直接掺入膳食食物。为了口服治疗施用,可以将活性化合物与赋形剂掺合并且以可摄入片剂、口含或舌下片、锭剂、胶囊、酏剂、混悬液、糖浆剂、糯米纸囊剂等形式使用。活性化合物在这类治疗上有用的组合物中的量是将得到适合剂量的量。
[0207]
所述片剂、锭剂、丸剂、胶囊等还可以包含如下文所列出的成分:粘合剂,例如树胶、阿拉伯胶、玉米淀粉或明胶;赋形剂,例如磷酸二钙;崩解剂,例如玉米淀粉、马铃薯淀粉、藻酸等;润滑剂,例如硬脂酸镁;以及甜味剂,例如蔗糖、乳糖或糖精,或矫味剂,例如薄荷、冬青油或樱桃香精。当剂型是胶囊时,除上述类型的物质外,它还可以包含液体载体。各种其它材料可以作为包衣衣料提供,否则可以改变剂型的物理形式。例如,可以用虫胶、糖或它们两者给片剂、丸剂或胶囊包衣。糖浆剂或酏剂可以包含活性化合物、作为甜味剂的蔗糖、作为防腐剂的对羟基苯甲酸甲酯和对羟基苯甲酸丙酯、染料和矫味剂例如樱桃或橙子香精。当然,用于制备任意单位剂型的任意材料应当是药学纯的且在使用量上基本上是无毒的。此外,可以将本发明的化合物掺入缓释制品和制剂,包括能够将活性肽特异性地递送至肠的特定区域的那些。
[0208]
还可以通过胃管或食道管经肠施用液体制剂。
[0209]
在一个实施方案中,将本发明的化合物与食物一起经口施用以促进转运至肠淋巴。
[0210]
在另一个实施方案中,与食物共同施用或不与食物共同施用,将本发明的化合物与基于脂质的制剂一起经口施用以促进转运至肠淋巴。
[0211]
用于经口递送的基于脂质的制剂是本领域中已知的且可以包括,例如基本上非水的介质,其典型地包含一种或多种脂质成分。脂质介质和得到的脂质制剂可以有用地如下所述根据其共有公同特征,按照脂质制剂分类系统(lfcs)进行分类(pouton,c.w.,eur.j.pharm.sci.11(增刊2),s93-s98,2000;pouton,c.w.,eur.j.pharm.sci.29 278-287,2006)。
[0212]
因此,脂质介质和得到的脂质制剂可以包含油/脂质和/或表面活性剂,任选地含有助溶剂。i型制剂包括需要消化的油或脂质,例如单甘油酯、二甘油酯和三甘油酯类及其组合。ii型制剂是不溶于水的自乳化递药系统(sedds),其包含用于i型制剂中的脂质和油以及另外的不溶于水的表面活性剂。iii型制剂为sedds或自微乳化递药系统(smedds),其包含用于i型制剂中的脂质和油以及另外的水溶性表面活性剂和/或助溶剂(iiia型)或更大比例的水溶性成分(iiib型)。iv型制剂主要包含亲水性表面活性剂和助溶剂(例如peg、丙二醇和二甘醇一乙醚)并且可用于难溶于水但非亲脂性的药物。本技术中包括任意这类
脂质制剂(i-iv型)。
[0213]
在一些实施方案中,脂质介质包含一种或多种油或脂质,不含额外的表面活性剂、辅助表面活性剂或辅助乳化剂或助溶剂,也就是说,它主要由一种或多种油或脂质组成。在另外一些实施方案中,脂质介质包含一种或多种油或脂质与一种或多种不溶于水的表面活性剂,任选地连同一种或多种助溶剂。在另外一些实施方案中,脂质介质包含一种或多种油或脂质与一种或多种水溶性表面活性剂,任选地连同一种或多种助溶剂。在一些实施方案中,脂质介质包含油/脂质、表面活性剂和助溶剂的混合物。在一些实施方案中,脂质介质基本上由一种或多种表面活性剂/辅助表面活性剂/辅助乳化剂和/或溶剂/助溶剂组成。
[0214]
可以用于本发明中的油或脂质的实例包括杏仁油、巴巴苏油、黑醋栗种子油、琉璃苣油、介花油、蓖麻油、椰子油、鳕鱼肝油、玉米油、棉籽油、月见草油、鱼油、葡萄籽油、芥菜籽油、橄榄油、棕榈仁油、棕榈油、花生油、菜籽油、红花油、芝麻油、鲨鱼肝油、大豆油、向日葵油、胡桃油、小麦胚芽油、鳄梨油、糠油、氢化蓖麻油、氢化椰子油、氢化棉籽油、氢化棕榈油、氢化大豆油、部分氢化大豆油、氢化植物油、辛酸/癸酸甘油三酯、分级分离的甘油三酯、三癸酸甘油酯、三己酸甘油酯、三辛酸甘油酯、三辛酸/癸酸甘油酯、三辛酸/癸酸甘油酯、三辛酸/癸酸/月桂酸甘油酯、三辛酸/癸酸/亚油酸甘油酯、三辛酸/癸酸/硬脂酸甘油酯、三月桂酸甘油酯、单月桂酸甘油酯、山嵛酸甘油酯、单亚油酸甘油酯、三亚油酸甘油酯、三油酸甘油酯、三十一酸甘油酯、甘油基三硬脂酸酯亚油酸甘油酯、饱和聚乙二醇化甘油酯、主要包含c
8-c
12
脂肪酸链的合成中链甘油三酯、主要包含c
8-c
12
脂肪酸链的中链甘油三酯、主要包含》c
12
脂肪酸链的长链甘油三酯、修饰的甘油三酯、分级分离的甘油三酯以及它们的混合物。
[0215]
可以用于本发明中的单酸甘油酯和甘油二酯的实例包括具有8-40个碳原子的脂肪酸链的一酯和二酯类,包括水解的椰子油(例如mcm)、水解的玉米油(例如maisine
tm
35-1)。在一些实施方案中,单酸甘油酯和甘油二酯是具有8-18个碳链长度的脂肪酸链的甘油的单饱和脂肪酸酯类或二饱和脂肪酸酯类(例如单硬脂酸甘油酯、二硬脂酸甘油酯、单辛酸甘油酯、二辛酸甘油酯、单癸酸甘油酯和二癸酸甘油酯)。
[0216]
用于脂质制剂中的适合表面活性剂包括c
8-c
22
脂肪酸的丙二醇一酯和二酯,例如,但不限于丙二醇单辛酸酯、丙二醇二辛酸酯、丙二醇单月桂酸酯,其以商品名例如90、pg、fcc销售;糖脂肪酸酯类,例如,但不限于棕榈酸蔗糖酯、月桂酸蔗糖酯、硬脂酸蔗糖酯;脱水山梨糖醇脂肪酸酯类,例如,但不限于月桂山梨坦、棕榈山梨坦、油酸山梨坦;聚氧乙烯水山梨糖醇脂肪酸酯类,例如,但不限于聚山梨醇酯20、聚山梨醇酯40、聚山梨醇酯60和聚山梨醇酯80、聚山梨醇酯85;聚氧乙烯一脂肪酸酯类和二脂肪酸酯类,包括,但不限于聚氧乙烯40硬脂酸酯和聚氧乙烯40油酸酯;c
8-c
22
脂肪酸的聚氧乙烯一酯类和二酯类的混合物和c
8-c
22
脂肪酸的甘油一酯类、二酯类和三酯类,其销售商品名为例如44/14、50/13、聚氧乙烯蓖麻油化合物,例如,但不限于聚氧乙烯35蓖麻油、聚氧乙烯40氢化蓖麻油和聚氧乙烯60氢化蓖麻油,其作为商品名例如/kolliphor el、rh40、rh60销售;聚氧乙烯烷
基醚,包括,但不限于聚氧乙烯20十六烷基十八烷基醚和聚氧乙烯10油醚;dl-.α.-生育酚聚乙二醇琥珀酸酯,其可以作为商品名销售;甘油一酯、二酯和三酯;c
8-c
22
脂肪酸的甘油一酯、二酯和三酯;蔗糖一酯、二酯和三酯;磺基琥珀酸二辛酸钠;聚氧乙烯-聚氧丙烯共聚物,例如,但不限于泊洛沙姆124、泊洛沙姆188、泊洛沙姆407;c
8-c
22
脂肪醇的聚氧乙烯醚类,包括,但不限于聚氧乙烯月桂醇、聚氧乙烯鲸蜡醇、聚氧乙烯十八烷醇、聚氧乙烯油醇,其销售用的商品名为例如35、58、7898;或者它们的任意两种或更多种的混合物。
[0217]
辅助乳化剂或辅助表面活性剂可以用于制剂中。适合的辅助乳化剂或辅助表面活性剂可以是磷酸甘油酯;磷脂,例如卵磷脂或游离脂肪酸,其在室温下为液体,例如异硬脂酸、油酸、亚油酸、亚麻酸、棕榈酸、硬脂酸、月桂酸、癸酸、辛酸和己酸。
[0218]
适合的溶剂/助溶剂包括乙醇、丙二醇、聚乙二醇、二甘醇一乙醚和甘油。
[0219]
聚合物也可以用于制剂中以抑制药物沉淀或改变药物释放速率。已经证实一系列聚合物可以影响这些特性且它们是本领域技术人员众所周知的。适合的聚合物包括羟丙基甲基纤维素、羟丙基甲基纤维素乙酰基琥珀酸酯、其它纤维素衍生的聚合物例如甲基纤维素;聚(甲基)丙烯酸酯类,例如eudragit系列聚合物,包括eudragit e100、聚乙烯吡咯烷酮或例如warren等人mol.pharmaceutics 2013,10,2823-2848中所述的其它聚合物。
[0220]
可以对制剂进行特别地选择,以提供活性成分在胃肠(gi)道中的持续释放,以便控制吸收速率。许多不同的方法可以用于实现这些目标,包括应用在gi道中缓慢地分散/浸蚀的高熔点脂质或形成缓慢地浸蚀的基质。这些制剂可以采用大整体型剂型的形式或可以作为微粒或纳米粒基质提供,例如如mishra,handbook of encapsulation and controlled release,crc press,boca raton,(2016)isbn 978-1-4822-3234-9,wilson和crowley controlled release in oral drug delivery,springer,ny,isbn 978-1-4614-1004-1(2011)或wise,handbook of pharmaceutical controlled release technology,marcel dekker,ny,isbn 0-82467-0369-3(2000)中所述。
[0221]
制剂还可以包含本领域技术人员通常已知基于脂质的制剂中包括的物质,包括抗氧化剂,例如丁羟茴醚(bha)或丁羟甲苯(bht);以及固化剂,例如微孔二氧化硅,例如偏硅酸铝镁(neusilin)。
[0222]
在另一个实施方案中,所述的化合物可以与酶抑制剂一起经口共同施用以增加前药在胃肠道或肠细胞中的稳定性。在一些实施方案中,想到所述酶抑制剂抑制胰脂肪酶,其实例包括,但不限于alli和奥利司他。在其它实施方案中,想到所述酶抑制剂抑制细胞脂肪酶,例如一酰基甘油脂肪酶,其实例包括,但不限于jzl184(4-硝基苯基-4-[双(1,3-苯并间二氧杂环戊烯-5-基)(羟基)甲基]哌啶-1-甲酸酯)。
[0223]
尽管如上文所述的化合物或其药学上可接受的盐可以以单独的活性成分施用于受试者,但是其它活性成分与所述化合物一起施用属于本发明的范围。在一个或多个实施方案中,想到本发明的化合物的两种或更多种的组合施用于受试者。
[0224]
本发明还提供药物组合物,其包含治疗有效量的如上文所定义的化合物或其药学上可接受的盐与至少一种药学上可接受的载体或稀释剂。
[0225]
术语“组合物”预期包括活性成分与作为载体的包囊材料的制剂,得到胶囊,其中活性成分(与其它载体一起或不与其它载体一起)被载体包围。
[0226]
正如本领域技术人员易于理解的,药学上可接受的载体的性质取决于被治疗的病症和哺乳动物的性质。认为特定载体或递送系统的选择易于由本领域技术人员确定。在制备包含活性化合物的任意制剂的过程中,应当小心以确保化合物的活性不在该过程中被破坏,且该化合物能够达到其作用部位而不受破坏。在一些情形中,有必要通过本领域公知的方式保护化合物,例如,微囊化。
[0227]
本领域技术人员采用常规方法可以轻易地确定用于本发明化合物的适合制剂。在本领域中鉴定优选的ph范围和适合的赋形剂例如抗氧化剂是常规的。缓冲系统常用于提供期望范围的ph值,并且包括羧酸缓冲液,例如乙酸盐、柠檬酸盐、乳酸盐和琥珀酸盐。各种抗氧化剂可以利用于这类制剂中,包括酚类化合物,例如bht或维生素e、还原剂例如甲硫氨酸或亚硫酸盐和金属螯合剂例如edta。
[0228]
药学上可接受的介质和/或稀释剂包括任意和所有的溶剂、分散介质、包衣、抗菌剂和抗真菌剂、等渗剂和吸收延迟剂等。这类介质和试剂对于药物活性物质的应用是本领域中众所周知的。除了任意常用介质或试剂与活性成分不相容,其在治疗组合物中的应用被考虑。还可以将补充活性成分掺入组合物。
[0229]
还可以将所述化合物与一种或多种其它治疗剂以组合方式施用。该组合能够单独、依次或同时施用上文所述的化合物与其它活性成分。该组合物可以以药物组合物的形式提供。
[0230]
如本技术中所用,术语“组合”是指组合物或部件套装产品,其中上文所定义的组合伴侣可以以依赖性或独立方式施用或通过使用与差别量的组合伴侣的不同固定组合来施用,即同时或在不同时间点施用。然后,例如,可以将组合伴侣同时或按时间交错施用,对于部件套装产品的不同组成部分,其可以在不同的时间点和相同或不同时间间隔施用。组合中施用的组合伴侣的总量的比例可变,例如,以便符合被治疗的患者亚群的需求或单一患者的需求,该不同需求可以归因于该患者的年龄、性别、体重等。
[0231]
尤其有利的是配制单位剂型形式的组合物,其易于施用且剂量均匀。如本技术中所用的单位剂型是指适合于作为被治疗的哺乳动物受试者的单元剂量的物理分散单元;每个单元包含预定量的活性物质,经计算其与所需的药学上可接受的介质结合产生期望的治疗作用。用于本发明新的单位剂型的规格根据如下因素指定并且直接取决于如下因素:(a)活性物质的独特特征和所实现的特定治疗作用;以及(b)混合用于治疗具有病况的活受试者的疾病的活性物质领域中内在的限制,如本技术中详细描公开的,所述受试者中身体健康受损。
[0232]
如上所述,可以混合主要活性成分,以方便和有效地以治疗有效量与适合的药学上可接受的介质一起在单位剂型中施用。例如,单位剂型可以包含量为0.25μg-约2000mg的主要活性化合物。按照比例表示,活性化合物的存在量约为0.25μg-约2000mg/ml载体。在包含补充活性成分的组合物的情况中,可以参照所述成分的常用剂量和施用方式确定剂量。
[0233]
如本技术中所用,术语“有效量”是指当根据期望的施药方案施用时提供期望的治疗活性的化合物的量。施药可以1次进行或以数分钟或数小时间隔进行,或在这些期限的任意一个内连续进行。适合的剂量可以在约0.1纳克/千克体重至1克/千克体重/剂量的范围。典型剂量为1微克至1克/千克体重/剂量,例如1毫克至1克/千克体重/剂量。在一个实施方
案中,该剂量可以为1毫克至500毫克/千克体重/剂量。在另一个实施方案中,该剂量可以为1毫克至250毫克/千克体重/剂量。在另一个实施方案中,该剂量可以为1毫克至100毫克/千克体重/剂量,例如至多50毫克/千克体重/剂量。
[0234]
如本技术中所用,术语“治疗”涵盖动物,优选哺乳动物,更优选人的病症或疾病的任意治疗,且包括治疗其中睾酮水平增加为有益的任意疾病或障碍。如本技术中所用,术语“预防”涵盖动物,优选哺乳动物,更优选人的病症或疾病的预防,且包括预防其中睾酮水平增加为有益的任意疾病或障碍。
[0235]
除非另有要求,否则在本说明书和权利要求书的自始至终中的措词“包含”和变化形式例如“含有”或“包括”将被理解为暗示包含所述整体或步骤或整体或步骤组,但不排除任意其它整体或整体组。
[0236]
本说明书中涉及任意现有出版物(或来源于它的信息)或已知的任意材料,不被且不应被视为认可或承认或任意形式建议现有出版物(或来源于它的信息)或已知的材料构成本说明书所涉及的领域中的公知常识的组成部分。
[0237]
现在参照下列非限制性实施例描述本发明。下列实施例是通式(i)的代表,并且提供了用于制备本发明示例性化合物的详细方法。
[0238]
实施例1.由酸酐合成酸-甘油三酯
[0239]
4-((1,3-双(棕榈酰氧基)丙-2-基)氧基)-4-氧代丁酸(iii)
[0240][0241]
将琥珀酸酐(iv)(25.4mg,0.254mmol)和dmap(15.5mg,0.127mmol)加到甘油二酯ii(72.2mg,0.127mmol)在吡啶(0.5ml)、ch2cl2(0.5ml)和thf(0.5ml)中的溶液中,将该混合物在室温搅拌17小时。此时tlc分析显示存在未反应的甘油二酯,因此再加入琥珀酸酐(25.4mg,0.254mmol)和dmap(15.5mg,0.127mmol),将该反应体系在40℃再加热22小时。将该混合物冷却至室温,用乙酸乙酯(20ml)稀释,用1m hcl(10ml)和盐水(2
×
30ml)洗涤,干燥(mgso4),减压浓缩,得到粗产物。通过硅胶色谱法纯化(15%-20%-25%乙酸乙酯/己烷),得到酸-tg iii(77.0mg,91%),为无色固体。
[0242]1h nmr(400mhz,cdcl3)δ5.27(m,1h),4.30(dd,j=12.0,4.3hz,2h),4.15(dd,j=12.0,5.8hz,2h),2.72
–
2.61(m,4h),2.31(t,j=7.6hz,4h),1.66
–
1.54(m,4h),1.35
–
1.19(m,48h),0.87(t,j=6.9hz,6h)。
[0243]
4-((1,3-双(棕榈酰氧基)丙-2-基)氧基)-3/2-甲基-4-氧代丁酸(iii)
[0244][0245]
将4-(二甲氨基)吡啶(64.6mg,0.527mmol)加到甘油二酯ii(200mg,0.351mmol)和
甲基琥珀酸酐(iv)(101mg,0.882mmol)在吡啶/thf/ch2cl2(各2.5ml)中的溶液中,将该混合物在室温搅拌22小时。用乙酸乙酯(40ml)稀释该反应体系,用1m hcl(30ml)和盐水(3
×
30ml)洗涤,干燥(mgso4),减压浓缩,得到粗制酸-tg iii(~1:1区域异构体混合物,240mg,定量),为无色固体,未纯化将其用于下一个反应。
[0246]1h nmr(400mhz,cdcl3)δ5.31
–
5.24(m,1h),4.34
–
4.27(m,2h),4.21
–
4.10(m,2h),3.00
–
2.90(m,1h),2.82
–
2.72(m,1h),2.52
–
2.43(m,1h),2.37
–
2.28(m,4h),1.67
–
1.55(m,4h),1.46(d,j=7.0hz,3h),1.36
–
1.17(m,48h),0.88(t,j=6.8hz,6h)。注意:整合和多重性反映出存在区域异构体混合物。
[0247]
实施例2.酸-甘油三酯的合成,其中r5是甲基
[0248]
a)二棕榈酸2-((4-((叔丁基二苯基甲硅烷基)氧基)-2-甲基丁酰基)氧基)丙-1,3-二基酯(vi)
[0249][0250]
将4-(二甲氨基)吡啶(dmap,59.9mg,0.491mmol)和edc
·
hcl(235mg,1.23mmol)加到酸v(175mg,0.491mmol)和1,3-dg ii(293mg,0.515mmol)在ch2cl2(8ml)中的溶液中,将该混合物在室温搅拌16小时。用ch2cl2(20ml)稀释该反应体系,加入硅胶,减压浓缩该混合物。通过硅胶色谱法纯化(3%-10%乙酸乙酯/己烷),得到甘油三酯vi(406mg,91%),为无色油状物。
[0251]1h nmr(400mhz,cdcl3)δ7.67
–
7.63(m,4h),7.45
–
7.34(m,6h),5.25(m,1h),4.30
–
4.22(m,2h),4.15
–
4.08(m,2h),3.69(t,j=6.3hz,2h),2.75(m,1h),2.27(t,j=7.2hz,2h),2.25(t,j=7.2hz,2h),1.99(m,1h),1.64
–
1.55(m,5h),1.32
–
1.20(m,48h),1.14(d,j=7.2hz,3h),1.04(s,9h),0.88(t,j=7.0hz,6h)。
[0252]
b)二棕榈酸2-((4-羟基-2-甲基丁酰基)氧基)丙-1,3-二基酯(vii)
[0253][0254]
在0℃将氟化四丁基铵(tbaf,1.0m的thf溶液,810μl,0.810μmol)和乙酸(46.1ml,0.810mmol)加到tbdps醚vi(406mg,0.447μmol)在thf(20ml)中的溶液中,将该混合物在室温搅拌3小时。用水(30ml)稀释该反应体系,用乙酸乙酯(3
×
20ml)萃取,用盐水(30ml)洗涤有机萃取物,干燥(mgso4),减压浓缩,得到粗制产物。通过硅胶色谱法纯化(0%-25%乙酸乙酯/己烷),得到醇vii(137mg,46%),为无色固体。
[0255]1h nmr(400mhz,cdcl3)δ5.27(m,1h),4.39(dd,j=12.0,4.1hz,1h),4.32(dd,j=11.9,4.3hz,1h),4.16(dd,j=12.0,6.0hz,1h),4.12(dd,j=12.0,5.6hz,1h),3.74
–
3.63(m,2h),2.66(m,1h),2.316(t,j=7.6hz,2h),2.309(t,j=7.4hz,2h),1.91(m,1h),1.70
(m,1h),1.63
–
1.58(m,4h),1.34
–
1.25(m,48h),1.19(d,j=7.1hz,3h),0.88(t,j=6.9hz,6h)。
[0256]
c)二棕榈酸2-((2-甲基-4-氧代丁酰基)氧基)丙-1,3-二基酯(viii)
[0257][0258]
在0℃将氯铬酸吡啶(pcc,89.1mg,0.410mmol)加到醇vii(137mg,0.205mmol)和c盐(celite)(90mg)在ch2cl2(12ml)中的混悬液中,将该混合物在室温搅拌2.5小时。通过短硅胶垫过滤该反应体系,用50%乙酸乙酯/己烷洗脱,减压浓缩滤液,得到粗制醛viii(134mg,定量),为黄色油状物,其未经纯化而被使用。
[0259]1h nmr(400mhz,cdcl3)δ9.75(s,1h),5.27(m,1h),4.30(dd,j=12.0,4.2hz,2h),4.16
–
4.09(m,2h),2.99(m,1h),2.89(ddd,j=18.1,7.8,0.8hz,1h),2.55(ddd,j=18.0,5.5,0.9hz,1h),2.32(t,j=7.4hz,2h),2.30(t,j=7.4hz,2h),1.64
–
1.58(m,4h),1.33
–
1.25(m,48h),1.22(d,j=7.1hz,3h),0.88(t,j=7.0hz,6h)。
[0260]
d)4-((1,3-双(棕榈酰氧基)丙-2-基)氧基)-3-甲基-4-氧代丁酸(iii)
[0261][0262]
将高锰酸钾(65.4mg,0.410μmol)加到在丙酮(7.5ml)和水(2.5ml)中的醛viii(134mg,0.205μmol)中,将该混合物在室温搅拌19小时。用水(25ml)稀释该反应体系,用1m hcl酸化至ph 2,用ch2cl2(3
×
20ml)萃取水层。用盐水(30ml)洗涤合并的有机萃取物,干燥(mgso4),减压浓缩,得到粗制产物。通过硅胶色谱法纯化(10%-25%乙酸乙酯/己烷),得到酸iii(79.6mg,58%),为无色固体。
[0263]1h nmr(400mhz,cdcl3)δ5.27(m,1h),4.32
–
4.27(m,2h),4.18
–
4.12(m,2h),2.92(m,1h),2.78(dd,j=16.9,8.0hz,1h),2.46(dd,j=16.9,6.0hz,1h),2.304(t,j=7.6hz,2h),2.297(t,j=7.6hz,2h),1.62
–
1.56(m,4h),1.31
–
1.19(m,51h),0.88(t,j=6.8hz,6h)。
[0264]
实施例3.通式(iii)的化合物的合成,其中r3是乙缩醛自消除(asi)基团
[0265]
((((8r,9s,10r,13s,14s,17s)-10,13-二甲基-3-氧代-2,3,6,7,8,9,10,11,12,13,14,15,16,17-十四氢-1h-环戊并[a]菲-17-基)氧基)甲基)琥珀酸1,3-双(棕榈酰氧基)丙-2-基酯(12)
[0266][0267]
在0℃将磺酰氯(13.3μl,0.164mmol)在ch2cl2(1ml)中的溶液加到mtm醚ix(46.9mg,0.135mmol)在ch2cl2(2ml)中的溶液中,将该混合物在0℃搅拌30分钟,然后在室温再搅拌1小时。在n2气流中浓缩该反应体系,从甲苯(2
×
5ml)中共蒸发,减压浓缩。然后将粗制残余物再溶于甲苯(1.5ml),加到预先搅拌1小时的酸iii(50.0mg,0.0747mmol)和dbu(16.8μl,0.112mmol)在甲苯(1.5ml)中的溶液中,将该混合物在室温搅拌2小时。用ch2cl2(20ml)稀释该反应体系,用饱和nahco3水溶液(20ml)和盐水(20ml)洗涤有机相,干燥(mgso4),减压浓缩,得到粗制产物。通过硅胶色谱法纯化(含有0.5%et3n的10%-25%乙酸乙酯/己烷),得到化合物12(8.4mg,12%),为淡黄色油状物。
[0268]1h nmr(400mhz,cdcl3)δ5.73(s,1h),5.34
–
5.23(m,3h),4.30(dd,j=11.9,4.4hz,2h),4.15(dd,j=11.9,5.8hz,2h),3.54(dd,j=8.3,8.3hz,1h),2.65(s,4h),2.48
–
2.24(m,8h),2.09
–
1.99(m,2h),1.92
–
1.80(m,2h),1.74
–
1.36(m,9h),1.35
–
1.21(m,49h),1.19(s,3h),1.17
–
0.91(m,5h),0.88(t,j=6.9hz,6h),0.80(s,3h)。
[0269]
esi-hrms:计算值c
59h101o10
[m+h
+
]969.7389;测定值969.7409.
[0270]
4-((((8r,9s,10r,13s,14s,17s)-10,13-二甲基-3-氧代-2,3,6,7,8,9,10,11,12,13,14,15,16,17-十四氢-1h-环戊并[a]菲-17-基)氧基)甲基)2/3-甲基琥珀酸1-(1,3-双(棕榈酰氧基)丙-2-基)酯(13/14)
[0271][0272]
在0℃将磺酰氯(21.4μl,0.264mmol)在ch2cl2(1.5ml)中的溶液加到mtm醚ix(76.3mg,0.219mmol)在ch2cl2(3ml)中的溶液中,将该混合物在0℃搅拌30分钟,然后在室温再搅拌45分钟。在n2气流中浓缩该反应体系,从甲苯(2
×
5ml)中共蒸发,减压浓缩。然后将粗制残余物再溶于甲苯(2.5ml),加到预先搅拌45分钟的酸iii(99.4mg,0.146mmol)和dbu(32.8μl,0.220mmol)在甲苯(2.5ml)中的溶液中,将该混合物在室温搅拌3小时。用乙酸乙酯(20ml)稀释该反应体系,用饱和nahco3水溶液(2
×
20ml)和盐水(2
×
20ml)洗涤有机相,干燥(mgso4),减压浓缩,得到粗制产物。通过硅胶色谱法纯化(含有0.5%et3n的5%-15%乙酸乙酯/己烷),得到化合物13和14(1:1区域异构体混合物,74.2mg,52%),为淡黄色油状物。
[0273]1h nmr(400mhz,cdcl3)δ5.73(s,1h),5.38
–
5.21(m,3h),4.32
–
4.25(m,2h),4.20
–
4.10(m,2h),3.58
–
3.50(m,1h),2.98
–
2.87(m,1h),2.81
–
2.71(m,1h),2.47
–
2.24(m,9h),2.08
–
1.98(m,2h),1.92
–
1.80(m,2h),1.75
–
1.51(m,8h),1.50
–
1.20(m,53h),1.19(s,3h),1.17
–
0.89(m,5h),0.88(t,j=7.0hz,6h),0.80(s,1.5h),0.79(s,1.5h)。注意:除可能的非对映异构体外,整合和多重性还反映出存在1:1个区域异构体混合物。
[0274]
4-((((8r,9s,10r,13s,14s,17s)-10,13-二甲基-3-氧代-2,3,6,7,8,9,10,11,12,13,14,15,16,17-十四氢-1h-环戊并[a]菲-17-基)氧基)甲基)2-甲基琥珀酸1-(1,3-双(棕榈酰氧基)丙-2-基)酯(13)
[0275][0276]
如上所述由酸-tg iii的α-甲基区域异构体制备。
[0277]1h nmr(400mhz,cdcl3)δ5.72(s,1h),5.32
–
5.22(m,3h),4.29/4.27(各自dd,j=12.0,4.2hz,2h),4.17/4.14(各自dd,j=11.9,6.0hz,2h),3.529/3.524(各自t,j=8.3hz,1h),2.93(m,1h),2.75(dd,j=16.8,7.9hz,1h),2.49
–
2.23(m,9h),2.08
–
1.96(m,2h),1.91
–
1.79(m,2h),1.71(m,1h),1.67
–
1.51(m,8h),1.49
–
1.07(m,51h),1.22(d,j=7.2hz,3h),1.18(s,3h),1.06
–
0.90(m,3h),0.87(t,j=6.8hz,6h),0.79(s,3h)。注意:双信号(例如:4.29/4.27)反映出存在非对映异构体混合物。
[0278]
esi-hrms:计算值c
60h102o10
na[m+na
+
]1005.7365;测定值1005.7370.
[0279]
实施例4.通式(iii)的化合物的合成,其中r3是羧基-乙缩醛或羧基-甲基乙缩醛自消除(casi或cmsi)基团
[0280]
a)((1s,4s)-4-(3,4-二氯苯基)-1,2,3,4-四氢萘-1-基)(甲基)氨基甲酸氯甲酯(xi)
[0281][0282]
在0℃将氯甲酸氯甲酯(25.9μl,0.292mmol)和吡啶(41.3μl,0.511mmol)加到在ch2cl2(4ml)中的盐酸舍曲林(50.0mg,0.146mmol)中,将该混合物在0℃搅拌15分钟,然后在室温搅拌1小时。用ch2cl2(20ml)稀释该反应体系,用饱和nahco3水溶液(20ml)和盐水(20ml)洗涤有机相,干燥(mgso4),减压浓缩,得到氨基甲酸氯甲酯xi(58.2mg,定量),为淡黄色油状物,其未经纯化而被使用。
[0283]1h nmr(400mhz,cdcl3)δ7.34(d,j=8.3hz,1h),7.31
–
7.25(m,1h),7.24
–
7.16(m,1h),7.11
–
7.06(m,1h),6.97(dd,j=7.0,2.0hz,1h),6.84
–
6.79(m,1h),5.91
–
5.83(m,1h),
5.51(dd,j=10.4,6.5hz,0.6h),5.32(dd,j=14.5,6.1hz,0.4h),4.20(dd,j=5.2,2.9hz,1h),2.78(s,1.2h),2.72(s,1.8h),2.36
–
2.22(m,1h),2.07
–
1.99(m,1h),1.87
–
1.70(m,2h)。注意:部分整合反映出存在旋转异构体混合物。
[0284]
b)(((((1s,4s)-4-(3,4-二氯苯基)-1,2,3,4-四氢萘-1-基)(甲基)氨基甲酰基)氧基)甲基)琥珀酸1,3-双(棕榈酰氧基)丙-2-基酯(1)
[0285][0286]
将1,8-二氮杂双环[5.4.0]十一-7-烯(dbu)(7.8μl,52.2μmol)加到酸-tg iii(25.1mg,37.5μmol)、氨基甲酸氯甲酯xi(13.0mg,32.6μmol)和碘化四丁基铵(tbai,3.6mg,9.8μmol)在甲苯(1.2ml)中的混悬液中,将该混合物回流加热1小时。将该反应体系冷却至室温,用乙酸乙酯(20ml)稀释,用水和盐水(各20ml)洗涤有机相,干燥(mgso4),减压浓缩,得到粗制产物。进行硅胶色谱法(15%乙酸乙酯/己烷),得到化合物1(29.0mg,86%),为无色油状物。
[0287]1h nmr(400mhz,cdcl3)δ7.33(d,j=8.3hz,1h),7.30
–
7.24(m,1h),7.22
–
7.17(m,2h),7.09(d,j=2.0hz,1h),6.96(d,j=7.3hz,1h),6.84
–
6.78(m,1h),5.89
–
5.83(m,2h),5.49(dd,j=10.1,6.6hz,0.6h),5.36
–
5.20(m,1.4h),4.33
–
4.25(m,2h),4.21
–
4.10(m,3h),2.78
–
2.61(m,7h),2.38
–
2.23(m,5h),2.06
–
1.96(m,1h),1.87
–
1.69(m,2h),1.67
–
1.53(m,4h),1.38
–
1.19(m,48h),0.88(t,j=6.9hz,6h)。注意:部分整合反映出存在旋转异构体混合物。
[0288]
esi-hrms:c
58h89
cl2no
10
na[m+na
+
]的计算值1052.5756;测定值1052.5774。
[0289]
a2)((1s,4s)-4-(3,4-二氯苯基)-1,2,3,4-四氢萘-1-基)(甲基)氨基甲酸1-氯乙酯(xi)
[0290][0291]
在0℃将氯甲酸1-氯乙酯(25.2μl,0.233mmol)和吡啶(35.4μl,0.438mmol)加到在ch2cl2(5ml)中的盐酸舍曲林(50.0mg,0.146mmol)中,将该混合物在0℃搅拌30分钟,然后在室温搅拌19小时。用ch2cl2(25ml)稀释该反应体系,用饱和nahco3水溶液(20ml)和盐水(20ml)洗涤有机相,干燥(mgso4),减压浓缩,得到粗制产物。通过硅胶色谱法纯化(含有0.5%et3n的15%乙酸乙酯/己烷),得到氨基甲酸氯乙酯xi(50.4mg,84%),为无色固体。
[0292]1h nmr(400mhz,cdcl3)δ7.34(d,j=8.3hz,1h),7.31
–
7.24(m,1h),7.24
–
7.16(m,2h),7.13
–
7.04(m,1h),7.00
–
6.94(m,1h),6.85
–
6.77(m,1h),6.75
–
6.64(m,1h),5.54
–
5.46(m,0.6h),5.41
–
5.33(m,0.4h),4.20(br s,1h),2.74/2.71/2.70(各自s,3h),2.36
–
2.24
(m,1h),2.09
–
1.97(m,1h),1.90
–
1.71(m,5h)。注意:部分整合和双信号反映出存在旋转异构体和非对映异构体二者。
[0293]
b2)(1-((((1s,4s)-4-(3,4-二氯苯基)-1,2,3,4-四氢萘-1-基)(甲基)氨基甲酰基)氧基)乙基)琥珀酸1,3-双(棕榈酰氧基)丙-2-基酯(2)
[0294][0295]
将1,8-二氮杂双环[5.4.0]十一-7-烯(dbu)(7.8μl,52.0μmol)加到酸-tg iii(25.0mg,37.4μmol)、氨基甲酸1-氯乙酯xi(13.4mg,32.5μmol)和碘化四丁基铵(tbai,3.6mg,9.7μmol)在甲苯(1.2ml)中的混悬液中,将该混合物回流加热1小时。将该反应体系冷却至室温,用乙酸乙酯(30ml)稀释,用水和盐水(各20ml)洗涤有机相,干燥(mgso4),减压浓缩,得到粗制产物。进行硅胶色谱法(4%-8%乙酸乙酯/甲苯),得到化合物2(19.7mg,58%),为无色油状物。
[0296]1h nmr(400mhz,cdcl3)δ7.33(d,j=8.3hz,1h),7.31
–
7.16(m,3h),7.12
–
7.06(m,1h),6.98
–
6.87(m,2h),6.85
–
6.77(m,1h),5.51
–
5.43(m,0.6h),5.36
–
5.16(m,1.4h),4.35
–
4.24(m,2h),4.21
–
4.11(m,3h),2.77
–
2.56(m,7h),2.37
–
2.24(m,5h),2.06
–
1.95(m,1h),1.85
–
1.71(m,2h),1.65
–
1.49(m,7h),1.37
–
1.19(m,48h),0.88(t,j=6.8hz,6h)。注意:部分整合和双信号反映出存在旋转异构体和非对映异构体二者。
[0297]
esi-hrms:c
59h91
cl2no
10
na[m+na
+
]的计算值1066.5912;测定值1066.5957。
[0298]
根据上述方法制得了下列包含羧基-乙缩醛或羧基-甲基乙缩醛自消除基团的化合物:
[0299]
a3)((4as,6r,7r,7ar,12bs)-3-(环丙基甲基)-6-((s)-2-羟基-3,3-二甲基丁-2-基)-7-甲氧基-1,2,3,4,5,6,7,7a-八氢-4a,7-桥亚乙基-4,12-亚甲基苯并呋喃并[3,2-e]异喹啉-9-基)碳酸氯甲酯(xi)
[0300][0301]1h nmr(400mhz,cdcl3):δ6.88(d,j=8.2hz,1h),6.61(d,j=8.2hz,1h),5.88(s,1h),5.82(d,j=6.3hz,1h),5.72(d,j=6.3hz,1h),4.47(d,j=1.7hz,1h),3.48(s,3h),3.06
–
2.98(m,2h),2.89(m,1h),2.63(dd,j=11.9,5.0hz,1h),2.40
–
2.23(m,4h),2.12(t,j=9.9hz,1h),2.03
–
1.79(m,3h),1.71(dd,j=12.9,2.5hz,1h),1.35(s,3h),1.28(m,2h),1.06(m,1h),1.03(s,9h),0.81(m,1h),0.66(m,1h),0.57
–
0.41(m,2h),0.16
–
0.08(m,2h)。
[0302]
b3)((((((4as,6r,7r,7ar,12bs)-3-(环丙基甲基)-6-((s)-2-羟基-3,3-二甲基丁-2-基)-7-甲氧基-1,2,3,4,5,6,7,7a-八氢-4a,7-桥亚乙基-4,12-亚甲基苯并呋喃并[3,2-e]异喹啉-9-基)氧基)羰基)氧基)甲基)琥珀酸1,3-双(棕榈酰氧基)丙-2-基酯(4)
[0303][0304]
如下所述,使用上述对化合物1和2所用方法的可替代方法制得了化合物4。将碳酸银(3.1mg,11.2μmol)加到在dmf(0.6ml)中的酸-tg iii(12.9mg,19.3μmol)中,将该混合物在室温搅拌1小时。减压浓缩该反应体系,得到灰色残余物,向其中加入在甲苯(0.6ml)和tbai(1.8mg,4.8μmol)中的碳酸氯甲酯xi(9.0mg,16.1μmol),将该混合物回流加热1.5小时。将该反应体系冷却至室温,然后用乙酸乙酯(30ml)稀释。用水(25ml)和盐水(25ml)洗涤有机相,干燥(mgso4),减压浓缩,得到粗制产物。进行硅胶色谱法(5%-15%乙酸乙酯/己烷),得到化合物4(3.7mg,19%),为无色固体。
[0305]1h nmr(400mhz,cdcl3)δ6.88(d,j=8.2hz,1h),6.60(d,j=8.1hz,1h),5.86(s,1h),5.82(s,2h),5.27(m,1h),4.46(s,1h),4.30(dd,j=12.0,4.3hz,2h),4.15(dd,j=12.0,5.9hz,2h),3.49(s,3h),3.06
–
2.98(m,2h),2.89(m,1h),2.73
–
2.60(m,5h),2.39
–
2.22(m,8h),2.12(t,j=9.2hz,1h),2.01(m,1h),1.93
–
1.80(m,2h),1.75
–
1.49(m,5h),1.35(s,3h),1.34
–
1.20(m,49h),1.05(m,1h),1.03(s,9h),0.88(t,j=6.9hz,6h),0.80(m,1h),0.66(m,1h),0.56
–
0.44(m,2h),0.16
–
0.09(m,2h)。
[0306]
esi-hrms:c
70h114
no
14
[m+h
+
]的计算值1192.8234;测定值1192.8244。
[0307]
(1-(((((4as,6r,7r,7ar,12bs)-3-(环丙基甲基)-6-((s)-2-羟基-3,3-二甲基丁-2-基)-7-甲氧基-1,2,3,4,5,6,7,7a-八氢-4a,7-桥亚乙基-4,12-亚甲基苯并呋喃并[3,2-e]异喹啉-9-基)氧基)羰基)氧基)乙基)琥珀酸1,3-双(棕榈酰氧基)丙-2-基酯(5)
[0308][0309]1h nmr(400mhz,cdcl3)δ6.88/6.87(各自d,j=8.2hz,1h),6.77(m,1h),6.59(d,j=8.2hz,1h),5.89/5.88(各自s,1h),5.25(m,1h),4.46(br s,1h),4.33
–
4.26(m,2h),
4.19
–
4.11(m,2h),3.49/3.48(各自s,3h),3.06
–
2.98(m,2h),2.89(m,1h),2.74
–
2.57(m,5h),2.38
–
2.20(m,8h),2.12(t,j=10.0hz,1h),1.98(td,j=12.5,5.5hz,1h),1.91
–
1.77(m,2h),1.70(dd,j=13.4,2.9hz,1h),1.64
–
1.52(m,7h),1.35(s,3h),1.33
–
1.18(m,49h),1.06(m,1h),1.03(s,5h),0.88(t,j=6.9hz,6h),0.81(m,1h),0.66(m,1h),0.55
–
0.43(m,2h),0.15
–
0.07(m,2h)。注意:双信号反映出存在非对映异构体混合物。
[0310]
esi-hrms:c
71h116
no
14
[m+h
+
]的计算值1206.8390;测定值1206.8401。
[0311]
(1-(((((8r,9s,10r,13s,14s,17s)-10,13-二甲基-3-氧代-2,3,6,7,8,9,10,11,12,13,14,15,16,17-十四氢-1h-环戊并[a]菲-17-基)氧基)羰基)氧基)乙基)琥珀酸1,3-双(棕榈酰氧基)丙-2-基酯(20)
[0312][0313]1h nmr(400mhz,cdcl3)δ6.75(m,1h),5.73(s,1h),5.25(m,1h),4.53(m,1h),4.34
–
4.26(m,2h),4.18
–
4.11(m,2h),2.73
–
2.58(m,4h),2.48
–
2.15(m,9h),2.02(m,1h),1.91
–
1.82(m,2h),1.78
–
1.55(m,8h),1.521(d,j=5.4hz,1.5h),1.517(d,j=5.4hz,1.5h),1.47
–
1.20(m,52h),1.19(s,3h),1.11
–
0.90(m,3h),0.88(t,j=7.5hz,6h),0.86(s,3h)。注意:双信号反映出存在非对映异构体混合物。
[0314]
esi-hrms:c
61h102o12
na[m+na
+
]的计算值1049.7263;测定值1049.7273。
[0315]
实施例5.通式(iii)的化合物的合成,其中r3是三甲基-锁(tml)自消除基团
[0316]
a)(2-(4-((叔丁基二甲基甲硅烷基)氧基)-2-甲基丁-2-基)-3,5-二甲基苯基)琥珀酸1,3-双(棕榈酰氧基)丙-2-基酯(xiv)
[0317][0318]
将4-(二甲氨基)吡啶(dmap,18.3mg,0.149mmol)和edc
·
hcl(71.6mg,0.374mmol)加到酸-tg iii(100mg,0.149mmol)和苯酚xiii(53.0mg,0.164mmol)在ch2cl2(4ml)中的溶液中,将该混合物在室温搅拌19小时。用ch2cl2(10ml)稀释该反应体系,加入硅胶,减压浓缩该混合物。通过硅胶色谱法纯化(3%-7.5%乙酸乙酯/己烷),得到tml甘油三酯xiv(84.6mg,58%),为无色油状物。
[0319]1h nmr(400mhz,cdcl3)δ6.80(d,j=2.0hz,1h),6.55(d,j=1.9hz,1h),5.29(m,1h),4.31(dd,j=11.9,4.4hz,2h),4.16(dd,j=12.0,5.8hz,2h),3.51
–
3.44(m,2h),2.85(t,j=6.9hz,2h),2.75(t,j=6.9hz,2h),2.51(s,3h),2.30(t,j=7.6hz,4h),2.22(s,3h),2.06
–
1.99(m,2h),1.65
–
1.56(m,4h),1.46(s,6h),1.37
–
1.20(m,48h),0.88(t,j=
6.9hz,6h),0.84(s,9h),
–
0.03(s,6h)。
[0320]
b)(2-(4-羟基-2-甲基丁-2-基)-3,5-二甲基苯基)琥珀酸1,3-双(棕榈酰氧基)丙-2-基酯(xv)
[0321][0322]
将10-樟脑磺酸(3.0mg,12.9μmol)加到在ch2cl2(1ml)和meoh(1ml)中的tbs醚xiv(83.7mg,86.0μmol)中,将该混合物在室温搅拌1小时。用水(10ml)稀释该反应体系,用ch2cl2(3
×
10ml)萃取水层。用饱和水溶液nahco3和盐水(各15ml)洗涤合并的有机萃取物,干燥(mgso4),减压浓缩,得到粗制产物。通过硅胶色谱法纯化(15%-25%乙酸乙酯/己烷),得到醇xv(59.9mg,81%),为无色油状物。
[0323]1h nmr(400mhz,cdcl3)δ6.81(d,j=2.0hz,1h),6.56(d,j=1.4hz,1h),5.28(m,1h),4.30(dd,j=12.0,4.4hz,2h),4.17(dd,j=12.0,5.8hz,2h),3.51(t,j=6.8hz,2h),2.88(t,j=6.6hz,2h),2.75(t,j=6.6hz,2h),2.52(s,3h),2.29(t,j=7.6hz,4h),2.22(s,3h),2.05(t,j=7.4hz,2h),1.65
–
1.57(m,4h),1.50(s,6h),1.37
–
1.20(m,48h),0.88(t,j=6.9hz,6h)。
[0324]
c)(3,5-二甲基-2-(2-甲基-4-氧代丁-2-基)苯基)琥珀酸1,3-双(棕榈酰氧基)丙-2-基酯(xvi)
[0325][0326]
在0℃将氯铬酸吡啶(pcc,30.1mg,0.139mmol)加到醇xv(59.9mg,0.0697mmol)和c盐(30mg)在ch2cl2(3ml)中的混悬液中,将该混合物在室温搅拌2小时。通过短硅胶垫过滤该反应体系,用50%乙酸乙酯/己烷洗脱,减压浓缩滤液,得到粗制醛xvi(59.8mg,定量),为黄色油状物,未经纯化使用它。
[0327]1h nmr(400mhz,cdcl3)δ9.54(t,j=2.6hz,1h),6.84(d,j=2.0hz,1h),6.60(d,j=1.4hz,1h),5.28(m,1h),4.30(dd,j=12.0,4.3hz,2h),4.16(dd,j=12.0,5.8hz,2h),2.86(t,j=6.7hz,2h),2.83(d,j=2.6hz,2h),2.75(t,j=6.3hz,2h),2.53(s,3h),2.30(t,j=7.6hz,4h),2.23(s,3h),1.64
–
1.58(m,4h),1.56(s,3h),1.55(s,3h),1.32
–
1.22(m,48h),0.88(t,j=6.9hz,6h)。
[0328]
d)1,3-(2-((4-((1,3-双(棕榈酰氧基)丙-2-基)氧基)-4-氧代丁酰基)氧基)-4,6-二甲基苯基)-3-甲基丁酸(xvii)
[0329][0330]
将高锰酸钾(12.2mg,76.7μmol)加到在丙酮(2.4ml)和水(0.8ml)中的醛xvi(59.8mg,69.7μmol)中,将该混合物在室温搅拌17小时。用水(10ml)稀释该反应体系,用1m hcl酸化至ph 2,用ch2cl2(3
×
15ml)萃取水层。用盐水(30ml)洗涤合并的有机萃取物,干燥(mgso4),减压浓缩,得到粗制产物。通过硅胶色谱法纯化(10%-25%乙酸乙酯/己烷),得到酸xvii(30.4mg,50%),为无色固体。
[0331]1h nmr(400mhz,cdcl3)δ6.81(d,j=1.6hz,1h),6.58(d,j=1.4hz,1h),5.28(m,1h),4.30(dd,j=11.9,4.4hz,2h),4.16(dd,j=12.0,5.8hz,2h),2.88(t,j=6.6hz,2h),2.84(s,2h),2.75(t,j=6.6hz,2h),2.53(s,3h),2.29(t,j=7.6hz,4h),2.22(s,3h),1.64
–
1.58(m,4h),1.57(s,6h),1.34
–
1.20(m,48h),0.88(t,j=6.8hz,6h)。
[0332]
e)(2-(4-(((8r,9s,10r,13s,14s,17s)-10,13-二甲基-3-氧代-2,3,6,7,8,9,10,11,12,13,14,15,16,17-十四氢-1h-环戊并[a]菲-17-基)氧基)-2-甲基-4-氧代丁-2-基)-3,5-二甲基苯基)琥珀酸1,3-双(棕榈酰氧基)丙-2-基酯(15)
[0333][0334]
将4-(二甲氨基)吡啶(dmap,4.1mg,33.2μmol)、edc
·
hcl(16.0mg,83.0μmol)和睾酮(17.2mg,60.0μmol)加到酸xvii(29.0mg,33.2μmol)在ch2cl2(1.2ml)中的溶液中,将该混合物在室温搅拌19小时。用ch2cl2(5ml)稀释该反应体系,加入硅胶,减压浓缩该混合物。通过硅胶色谱法纯化(12.5%-20%乙酸乙酯/己烷),得到化合物15(15.0mg,40%),为无色固体。
[0335]1h nmr(400mhz,cdcl3)δ6.80(d,j=2.0hz,1h),6.57(d,j=1.9hz,1h),5.72(s,1h),5.28(m,1h),4.46(dd,j=9.1,7.3hz,1h),4.31(dd,j=11.9,4.4hz,2h),4.16(dd,j=11.9,5.8hz,2h),2.88(t,j=6.7hz,2h),2.81(d,j=11.4hz,2h),2.76(t,j=6.9hz,2h),2.54(s,3h),2.50
–
2.23(m,8h),2.21(s,3h),2.14
–
1.96(m,2h),1.81(m,1h),1.69(m,1h),1.65
–
1.47(m,14h),1.40
–
1.20(m,51h),1.17(s,3h),1.10
–
0.92(m,4h),0.88(t,j=6.9hz,6h),0.66(s,3h)。
[0336]
esi-hrms:c
71h115o11
[m+h
+
]的计算值1143.8434;测定值1143.8443。
[0337]
根据上述方法制得了下列包含三甲基-锁自消除基团的化合物:
[0338]
(2-(4-(((1s,4s)-4-(3,4-二氯苯基)-1,2,3,4-四氢萘-1-基)(甲基)氨基)-2-甲基-4-氧代丁-2-基)-3,5-二甲基苯基)琥珀酸1,3-双(棕榈酰氧基)丙-2-基酯(3)
[0339][0340]
将4-(二甲氨基)吡啶(dmap,2.8mg,22.9μmol)、edc
·
hcl(11.0mg,57.3μmol)和三乙胺(8.0μl,57.3μmol)加到盐酸舍曲林(10.2mg,29.8μmol)和酸xvii(20.0mg,22.9μmol)在ch2cl2(1ml)中的溶液中,将该混合物在室温搅拌16小时。用ch2cl2(5ml)稀释该反应体系,加入硅胶,减压浓缩该混合物。通过硅胶色谱法纯化(15%-25%乙酸乙酯/己烷),得到化合物3(22.2mg,83%),为无色固体。
[0341]1h nmr(400mhz,cdcl3)δ7.32(d,j=8.3hz,0.7h),7.31(d,j=8.3hz,0.3h),7.25
–
7.11(m,2h),7.08
–
7.02(m,1.3h),6.97
–
6.85(m,1.7h),6.83
–
6.79(m,1.7h),6.73(dd,j=8.3,2.0hz,0.3h),6.60(d,j=1.8hz,0.7h),6.57(d,j=1.7hz,0.3h),5.88(dd,j=10.5,6.3hz,0.7h),5.24(m,1h),4.96(dd,j=10.9,5.7hz,0.3h),4.32
–
4.25(m,2h),4.20
–
4.10(m,3h),3.09(d,j=15.5hz,0.7h),3.01(d,j=8.2hz,0.3h),2.92
–
2.73(m,4.3h),2.70
–
2.64(m,3h),2.60
–
2.56(m,3.7h),2.29(t,j=7.5hz,4h),2.23(s,2.1h),2.21(s,0.9h),2.21(m,1h),1.95(m,1h),1.69(s,2.1h),1.66(s,0.9h),1.62(s,3h),1.70
–
1.52(m,6h),1.34
–
1.20(m,48h),0.88(t,j=6.9hz,6h)。注意:部分整合反映出存在旋转异构体混合物。
[0342]
esi-hrms:c
69h103
cl2no9na[m+na
+
]的计算值1182.6902;测定值1182.6904。
[0343]
(2-(4-(((4as,6r,7r,7ar,12bs)-3-(环丙基-甲基)-6-((s)-2-羟基-3,3-二甲基丁-2-基)-7-甲氧基-1,2,3,4,5,6,7,7a-八氢-4a,7-桥亚乙基-4,12-亚甲基苯并呋喃并[3,2-e]异喹啉-9-基)氧基)-2-甲基-4-氧代丁-2-基)-3,5-二甲基苯基)琥珀酸1,3-双(棕榈酰氧基)丙-2-基酯(6)
[0344][0345]1h nmr(400mhz,cdcl3)δ6.80(d,j=1.9hz,1h),6.61
–
6.57(m,2h),6.53(d,j=8.1hz,1h),5.90(s,1h),5.28(m,1h),4.39(s,1h),4.30(dd,j=11.9,4.4hz,2h),4.16(dd,j=11.9,5.8hz,2h),3.37(s,3h),3.06(abq,2h),3.01
–
2.81(m,5h),2.76(t,j=6.7hz,2h),2.60(dd,j=11.6,4.8hz,1h),2.55(s,3h),2.29(t,j=7.6hz,4h),2.37
–
2.18(m,4h),2.21(s,3h),2.10(t,j=9.8hz,1h),1.95(td,j=12.6,5.4hz,1h),1.89
–
1.74(m,2h),
1.71
–
1.51(m,11h),1.33(s,3h),1.45
–
1.14(m,49h),1.02(s,9h),0.88(t,j=6.9hz,6h),0.83
–
0.61(m,2h),0.54
–
0.42(m,2h)。0.14
–
0.07(m,2h)。
[0346]
esi-hrms:c
81h128
no
13
[m+h
+
]的计算值1322.9380;测定值1322.9404。
[0347]
实施例6.通式(iii)的化合物的合成,其中r3是对羟基苄基(phb)羰基自消除基团
[0348]
a)(4-(((叔丁基二甲基甲硅烷基)氧基)甲基)-苯基)琥珀酸1,3-双(棕榈酰氧基)丙-2-基酯(xxi)
[0349][0350]
将4-(二甲氨基)吡啶(dmap,48.0mg,0.393mmol)和edc
·
hcl(123mg,0.639mmol)加到酸-tg iii(200mg,0.300mmol)和苯酚xx(93.1mg,0.391mmol)在ch2cl2(15ml)中的溶液中,将该混合物在室温搅拌21小时。用ch2cl2(15ml)稀释该反应体系,加入硅胶,减压浓缩该混合物。通过硅胶色谱法纯化(5%-15%乙酸乙酯/己烷),得到phb甘油三酯xxi(202mg,76%),为无色油状物。
[0351]1h nmr(400mhz,cdcl3)δ7.34
–
7.29(m,2h),7.07
–
7.02(m,2h),5.30(m,1h),4.72(s,2h),4.31(dd,j=12.0,4.3hz,2h),4.16(dd,j=12.0,5.9hz,2h),2.88(t,j=6.9hz,2h),2.76(t,j=6.5hz,2h),2.29(t,j=7.6hz,4h),1.63
–
1.55(m,4h),1.33
–
1.20(m,48h),0.94(s,9h),0.88(t,j=6.9hz,6h),0.09(s,6h)。
[0352]
b)(4-(羟基甲基)苯基)琥珀酸1,3-双(棕榈酰氧基)丙-2-基酯(xxii)
[0353][0354]
将10-樟脑磺酸(8.0mg,0.0344mmol)加到在ch2cl2(2.5ml)和meoh(2.5ml)中的tbs醚xxi(181mg,0.203mmol)中,将该混合物在室温搅拌3.5小时。用ch2cl2(30ml)稀释该反应体系,用饱和nahco3水溶液(2
×
20ml)和盐水(20ml)洗涤有机相,干燥(mgso4),减压浓缩,得到粗制产物。通过硅胶色谱法纯化(10%-25%乙酸乙酯/己烷),得到醇xxii(128mg,81%),为无色固体。
[0355]1h nmr(400mhz,cdcl3)δ7.40
–
7.35(m,2h),7.11
–
7.06(m,2h),5.30(m,1h),4.69(d,j=5.9hz,2h),4.31(dd,j=12.0,4.3hz,2h),4.16(dd,j=12.0,5.9hz,2h),2.92
–
2.86(m,2h),2.79
–
2.73(m,2h),2.29(t,j=7.6hz,4h),1.64
–
1.55(m,4h),1.34
–
1.19(m,48h),0.88(t,j=6.9hz,6h)。
[0356]
c)(4-((((4-硝基苯氧基)羰基)氧基)甲基)-苯基)琥珀酸1,3-双(棕榈酰氧基)丙-2-基酯(xxiii)
[0357][0358]
在0℃将氯甲酸4-硝基苯基酯(8.4mg,41.6μmol)和吡啶(3.8μl,47.0μmol)加到在ch2cl2(2ml)中的醇xxii(20.0mg,25.8μmol)中,将该混合物在0℃搅拌30分钟,然后在室温搅拌4.5小时。用ch2cl2(30ml)稀释该反应体系,用饱和nahco3水溶液和盐水(各3
×
25ml)洗涤有机相,干燥(mgso4),减压浓缩,得到粗制产物。通过硅胶色谱法纯化(10%-20%乙酸乙酯/己烷),得到碳酸pnp酯xxiii(15.7mg,65%),为无色固体。
[0359]1h nmr(400mhz,cdcl3)δ8.31
–
8.25(m,2h),7.48
–
7.43(m,2h),7.41
–
7.36(m,2h),7.17
–
7.11(m,2h),5.29(m,1h),5.28(s,2h),4.32(dd,j=12.0,4.3hz,2h),4.17(dd,j=12.0,5.8hz,2h),2.93
–
2.87(m,2h),2.79
–
2.73(m,2h),2.30(t,j=7.6hz,4h),1.63
–
1.51(m,4h),1.34
–
1.18(m,48h),0.88(t,j=6.9hz,6h)。
[0360]
d)(4-((((((8r,9s,10r,13s,14s,17s)-10,13-二甲基-3-氧代-2,3,6,7,8,9,10,11,12,13,14,15,16,17-十四氢-1h-环戊丙[a]菲-17-基)氧基)羰基)氧基)甲基)苯基)琥珀酸1,3-双(棕榈酰氧基)丙-2-基酯(16)
[0361][0362]
将4-(二甲氨基)吡啶(dmap,21.3mg,0.174mmol)和dipea(7.1μl,0.0406mmol)加到睾酮(48.0mg,0.166mmol)和碳酸pnp酯xxiii(127mg,0.135mmol)在ch2cl2(10ml)中的溶液中,将该混合物在室温搅拌5天。用ch2cl2(20ml)稀释该反应体系,用1m hcl、水和盐水(各20ml)洗涤,干燥(mgso4),减压浓缩,得到粗制产物。通过硅胶色谱法纯化(5%乙酸乙酯/甲苯),得到化合物16(20.4mg,14%),为无色固体。
[0363]1h nmr(400mhz,cdcl3)δ7.42
–
7.37(m,2h),7.12
–
7.05(m,2h),5.73(s,1h),5.29(m,1h),5.12(s,2h),4.52(t,j=8.4hz,1h),4.31(dd,j=12.0,4.3hz,2h),4.16(dd,j=12.0,5.9hz,2h),2.89(t,j=6.7hz,2h),2.76(t,j=6.7hz,2h),2.47
–
2.34(m,3h),2.29(t,j=7.6hz,4h),2.34
–
2.16(m,2h),2.02(m,1h),1.89
–
1.80(m,2h),1.74
–
1.53(m,8h),1.47
–
1.19(m,51h),1.18(s,3h),1.10
–
0.92(m,4h),0.88(t,j=6.9hz,6h),0.85(s,3h)。
[0364]
esi-hrms:c
66h105o12
[m+h
+
]的计算值1089.7601;测定值1089.7617。
[0365]
实施例7.通式(iii)的化合物的合成,其中r3是翻转酯自消除(fsi)基团
[0366]
a)4-溴丁酸(8r,9s,10r,13s,14s,17s)-10,13-二甲基-3-氧代-2,3,6,7,8,9,10,11,12,13,14,15-16,17-十四氢-1h-环戊并[a]菲-17-基酯(xxvi)
[0367][0368]
将4-(二甲氨基)吡啶(dmap,15.5mg,0.130mmol)和dcc(43.8mg,0.210mmol)加到睾酮(29.9mg,0.100mmol)和4-溴丁酸(xxv)(21.0mg,0.130mmol)在ch2cl2(3ml)中的溶液中,将该混合物在室温搅拌24小时。再加入0.6eq.酸、1eq.dcc、0.6eq.dmap,将该混合物在室温再搅拌2天。用ch2cl2(10ml)稀释该反应体系,加入硅胶,减压浓缩该混合物。通过硅胶色谱法纯化(25%乙酸乙酯/己烷),得到溴化物xxvi(26.7mg,59%),为无色固体。
[0369]1h nmr(400mhz,cdcl3)δ5.73(s,1h),4.62(dd,j=9.1,7.9hz,1h),3.47(t,j=6.5hz,2h),2.50(td,j=7.1,1.0hz,2h),2.47
–
2.23(m,4h),2.22
–
2.13(m,3h),2.06
–
1.99(m,1h),1.85(m,1h),1.78(m,1h),1.74
–
1.63(m,2h),1.61
–
1.53(m,2h),1.52
–
1.32(m,3h),1.23
–
1.15(m,1h),1.19(s,3h)1.11
–
0.91(m,3h),0.83(s,3h)。
[0370]
b)(4-(((8r,9s,10r,13s,14s,17s)-10,13-二甲基-3-氧代-2,3,6,7,8,9,10,11,12,13,14,15,16,17-十四氢-1h-环戊并[a]菲-17-基)氧基)-4-氧代丁基)琥珀酸1,3-双(棕榈酰氧基)丙-2-基酯(17)
[0371][0372]
将1,8-二氮杂双环[5.4.0]十一-7-烯(dbu)(9.9μl,66.2μmol)加到酸-tg iii(30.9mg,46.2μmol)和溴化物xxvi(18.3mg,41.8μmol)在甲苯(1.5ml)中的混悬液中,将该混合物回流加热21小时。将该反应体系冷却至室温,然后用乙酸乙酯(20ml)稀释。用水(10ml)和盐水(10ml)洗涤有机相,干燥(mgso4),减压浓缩,得到粗制产物。进行硅胶色谱法(15%-25%乙酸乙酯/己烷),得到化合物17(21.6mg,50%),为无色固体。
[0373]1h nmr(400mhz,cdcl3)δ5.73(s,1h),5.26(m,1h),4.62(dd,j=9.1,7.9hz,1h),4.30(dd,j=11.9,4.4hz,2h),4.15(dd,j=11.9,5.8hz,2h),4.13(t,j=6.5hz,2h),2.69
–
2.58(m,4h),2.47
–
2.25(m,9h),2.19(m,1h),2.07
–
1.91(m,3h),1.85(m,1h),1.78(m,1h),1.75
–
1.45(m,9h),1.44
–
1.21(m,59h),1.19(s,3h),1.16
–
0.91(m,5h),0.88(t,j=6.9hz,6h),0.83(s,3h)。
[0374]
esi-hrms:c
62h104o11
na[m+na
+
]的计算值1047.7471;测定值1047.7460。
[0375]
根据上述方法制得了下列包含翻转酯自消除基团的化合物:
[0376]
(5-(((8r,9s,10r,13s,14s,17s)-10,13-二甲基-3-氧代-2,3,6,7,8,9,10,11,12,13,14,15,16,17-十四氢-1h-环戊并[a]菲-17-基)氧基)-5-氧代戊基)琥珀酸1,3-双(棕榈酰氧基)丙-2-基酯(18)
[0377][0378]1h nmr(400mhz,cdcl3)δ5.73(s,1h),5.26(m,1h),4.61(dd,j=9.1,7.9hz,1h),4.29(dd,j=11.9,4.4hz,2h),4.15(dd,j=11.9,5.8hz,2h),4.10(t,j=6.0hz,2h),2.68
–
2.58(m,4h),2.47
–
2.25(m,10h),2.18(m,1h),2.02(ddd,j=13.3,4.9,3.3hz,1h),1.85(m,1h),1.77(m,1h),1.74
–
1.45(m,13h),1.44
–
1.21(m,50h),1.19(s,3h),1.18
–
0.91(m,4h),0.88(t,j=6.9hz,6h),0.83(s,3h)。
[0379]
esi-hrms:c
63h107o11
na[m+na
+
]的计算值1061.7627;测定值1061.7654。
[0380]
4-(5-(((8r,9s,10r,13s,14s,17s)-10,13-二甲基-3-氧代-2,3,6,7,8,9,10,11,12,13,14,15,16,17-十四氢-1h-环戊并[a]菲-17-基)氧基)-5-氧代戊基)2-甲基琥珀酸1-(1,3-双(棕榈酰氧基)丙-2-基)酯(19)
[0381][0382]1h nmr(400mhz,cdcl3)δ5.73(s,1h),5.26(m,1h),4.61(dd,j=9.1,7.9hz,1h),4.33
–
4.24(m,2h),4.21
–
4.04(m,4h),2.91(m,1h),2.76/2.72(各自dd,j=13.9,7.9hz,1h),2.47
–
2.26(m,11h),2.18(m,1h),2.02(m,1h),1.84(m,1h),1.77(m,1h),1.74
–
1.44(m,13h),1.43
–
1.14(m,51h),1.223/1.214(各自d,j=7.2hz,3h),1.19(s,3h),1.11
–
0.92(m,3h),0.88(t,j=6.9hz,6h),0.83(s,3h)。注意:双信号反映出存在非对映异构体混合物。
[0383]
esi-hrms:c
64h109o11
[m+h
+
]的计算值1053.7964;测定值1053.7984。
[0384]
(5-(((4as,6r,7r,7ar,12bs)-3-(环丙基甲基)-6-((s)-2-羟基-3,3-二甲基丁-2-基)-7-甲氧基-1,2,3,4,5,6,7,7a-八氢-4a,7-桥亚乙基-4,12-亚甲基苯并呋喃并[3,2-e]异喹啉-9-基)氧基)-5-氧代戊基)琥珀酸1,3-双(棕榈酰氧基)丙-2-基酯(7)
[0385][0386]1h nmr(400mhz,cdcl3)δ6.77(d,j=8.1hz,1h),6.59(d,j=8.1hz,1h),5.88(s,1h),5.26(m,1h),4.42(d,j=1.6hz,1h),4.29(dd,j=11.9,4.4hz,2h),4.15(dd,j=12.0,5.8hz,2h),4.11(t,j=6.0hz,2h),3.45(s,3h),3.02(d,j=13.5hz,1h),2.99(s,1h),2.88(m,1h),2.67
–
2.54(m,7h),2.38
–
2.22(m,8h),2.11(t,j=10.1hz,1h),1.97(td,j=12.7,5.6hz,1h),1.91
–
1.84(m,2h),1.82
–
1.68(m,5h),1.65
–
1.57(m,4h),1.35(s,3h),1.35
–
1.19(m,49h),1.05(m,1h),1.03(s,9h),0.88(t,j=6.9hz,6h),0.84
–
0.76(m,1h),0.75
–
0.63(m,1h),0.55
–
0.43(m,2h),0.17
–
0.08(m,2h)。
[0387]
esi-hrms:c
73h120
no
13
[m+h
+
]的计算值1218.8754;测定值1218.8775。
[0388]
4-(5-(((4as,6r,7r,7ar,12bs)-3-(环丙基甲基)-6-((s)-2-羟基-3,3-二甲基丁-2-基)-7-甲氧基-1,2,3,4,5,6,7,7a-八氢-4a,7-桥亚乙基-4,12-亚甲基苯并呋喃并[3,2-e]异喹啉-9-基)氧基)-5-氧代戊基)2-甲基琥珀酸1-(1,3-双(棕榈酰氧基)丙-2-基)酯(8)
[0389][0390]1h nmr(400mhz,cdcl3)δ6.77(d,j=8.1hz,1h),6.59(d,j=8.1hz,1h),5.88(s,1h),5.26(m,1h),4.42(d,j=1.5hz,1h),4.32
–
4.24(m,2h),4.20
–
4.06(m,4h),3.45(s,3h),3.02(d,j=13.3hz,1h),2.99(s,1h),2.96
–
2.84(m,2h),2.74(ddd,j=16.6,13.4,8.0hz,1h),2.66
–
2.53(m,3h),2.46
–
2.21(m,9h),2.11(t,j=9.9hz,1h),1.97(td,j=12.5,5.6hz,1h),1.92
–
1.67(m,7h),1.65
–
1.56(m,4h),1.35(s,3h),1.34
–
1.17(m,49h),1.222(d,j=7.2hz,1.5h),1.116(d,j=7.2hz,1.5h),1.05(m,1h),1.03(s,9h),0.88(t,j=6.8hz,6h),0.80(m,1h),0.67(m,1h),0.55
–
0.42(m,2h),0.15
–
0.08(m,2h)。
[0391]
esi-hrms:c
74h122
no
13
[m+h
+
]的计算值1232.8911;测定值1232.8925。
[0392]
(5-((1-(异丙基氨基)-3-(4-(2-甲氧基乙基)-苯氧基)丙-2-基)氧基)-5-氧代戊基)琥珀酸1,3-双(棕榈酰氧基)丙-2-基酯(9)
[0393][0394]
在0℃将三氟乙酸(tfa,6.1μl,82.2μmol)加到在ch2cl2(1.2ml)中的boc-保护的前药xxvii(9.2mg,8.2μmol)中,将该混合物在室温搅拌21小时。在n2气流中浓缩该反应体系,得到粗制产物。进行硅胶色谱法(1%-3.5%甲醇/ch2cl2),得到化合物9(6.8mg,81%),为淡黄色油状物。
[0395]1h nmr(400mhz,cdcl3)δ7.13(d,j=8.6hz,2h),6.84
–
6.77(m,2h),5.38(m,1h),5.24(m,1h),4.28(dd,j=11.9,4.5hz,2h),4.18
–
4.10(m,4h),4.06(t,j=5.7hz,2h),3.55(t,j=7.0hz,2h),3.34(s,3h),3.38
–
3.24(m,3h),2.81(t,j=7.0hz,2h),2.66
–
2.56(m,4h),2.48
–
2.35(m,2h),2.31(t,j=7.6hz,4h),1.69
–
1.54(m,8h),1.32(d,j=6.4hz,6h),1.37
–
1.19(m,48h),0.88(t,j=6.9hz,6h)。
[0396]
esi-hrms:c
59h104
no
12
[m+h
+
]的计算值1018.7553;测定值1018.7568。
[0397]
实施例8.通式(iv)的化合物的合成,其中药用物质是麦考酚酸(mpa)
[0398]
a)5-(氯甲基)3-甲基戊二酸1-(1,3-双(棕榈酰氧基)丙-2-基)酯(xviii)
[0399][0400]
将酸-tg iii(75.0mg,0.108mmol)、n,n-二甲基甲酰胺(dmf,1滴)和socl2(78.0μl,1.08mmol)的混合物回流加热45分钟,然后冷却至室温。减压浓缩该反应体系,然后从甲苯(各3ml)中共蒸发3次,减压干燥。将得到的酰氯再溶于ch2cl2(1ml),滴加到在ch2cl2(0.5ml)中的无水zrcl4(25.1mg,0.108mmol)中,在室温搅拌15分钟,然后冷却至0℃。加入1,3,5-三烷(9.7mg,0.108mmol),将该混合物在室温搅拌20小时。用ch2cl2(15ml)稀释该反应体系,用水和盐水(各15ml)洗涤有机相,干燥(mgso4),减压浓缩,得到粗制产物。通过硅胶色谱法纯化(5%-12.5%乙酸乙酯/己烷),得到氯甲酯xxviii(19.2mg,24%),为黄色油状物。
[0401]1h nmr(400mhz,cdcl3)δ5.72
–
5.67(m,2h),5.27(m,1h),4.31(dd,j=11.9,4.2hz,2h),4.13(dd,j=12.0,6.1hz,2h),2.53
–
2.34(m,5h),2.31(t,j=7.6hz,4h),1.67
–
1.53(m,4h),1.37
–
1.19(m,48h),1.05(d,j=6.5hz,3h),0.88(t,j=6.9hz,6h)。
[0402]
b)3-甲基戊二酸(e)-1-(((6-(4-(烯丙氧基)-6-甲氧基-7-甲基-3-氧代-1,3-二氢异苯并呋喃-5-基)-4-甲基己-4-烯酰基)氧基)甲基)5-(1,3-双(棕榈酰氧基)丙-2-基)酯(xxxi)
[0403][0404]
将1,8-二氮杂双环[5.4.0]十一-7-烯(dbu)(6.9μl,45.9μmol)加到mpa(oall)xxx(12.9mg,35.7μmol)和氯甲基酯xxviii(19.0mg,25.5μmol)和tbai(4.7mg,12.7μmol)在甲苯(0.8ml)中的混悬液中,将该混合物在80℃加热2小时。将该反应体系冷却至室温,然后用乙酸乙酯(20ml)稀释。用水和盐水(各20ml)洗涤有机相,干燥(mgso4),减压浓缩,得到粗制产物。进行硅胶色谱法(15%-20%乙酸乙酯/己烷),得到被保护的前药xxxi(16.7mg,61%),为无色固体。
[0405]1h nmr(400mhz,cdcl3)δ6.09(m,1h),5.69(s,2h),5.36(m,1h),5.31
–
5.22(m,2h),5.18(td,j=6.7,1.2hz,1h),5.13(s,2h),4.78(dt,j=5.9,1.2hz,2h),4.292/4.288(各自dd,j=11.9,4.3hz,2h),4.13(dd,j=11.9,5.9hz,2h),3.76(s,3h),3.41(d,j=6.7hz,2h),2.50
–
2.37(m,5h),2.34
–
2.23(m,8h),2.18(s,3h),1.77(s,3h),1.64
–
1.56(m,4h),1.35
–
1.18(m,48h),1.02(d,j=6.4hz,3h),0.87(t,j=6.9hz,6h)。注意:双信号反映出存在非对映异构体混合物。
[0406]
c)3-甲基-戊二酸(e)-1-(1,3-双(棕榈酰氧基)丙-2-基)5-(((6-(4-羟基-6-甲氧基-7-甲基-3-氧代-1,3-二氢异苯并呋喃-5-基)-4-甲基己-4-烯酰基)氧基)甲基)酯(10)
[0407][0408]
将1,3-二甲基巴比妥酸(4.9mg,31.2μmol)和pd(pph3)4(5.4mg,4.7μmol)加到在ch2cl2(0.5ml)中的烯丙基醚xxxi(16.7mg,15.6μmol)中,将该混合物在室温搅拌2小时。将该反应混合物直接上短硅胶垫,用50%乙酸乙酯/己烷洗脱。减压浓缩洗脱液,得到粗制产物,通过硅胶色谱法纯化(5%-10%乙酸乙酯/甲苯),得到化合物10(10.1mg,63%),为无色固体。
[0409]1h nmr(400mhz,cdcl3)δ7.68(s,1h),5.70(s,2h),5.30
–
5.21(m,2h),5.20(s,2h),4.296/4.291(各自dd,j=11.9,4.3hz,2h),4.13(dd,j=11.9,6.0hz,2h),3.76(s,3h),3.38(d,j=6.9hz,2h),2.49
–
2.38(m,5h),2.34
–
2.25(m,8h),2.15(s,3h),1.79(s,3h),1.66
–
1.54(m,4h),1.35
–
1.19(m,48h),1.02(d,j=6.4hz,3h),0.88(t,j=6.9hz,6h)。注意:双信号反映出存在非对映异构体混合物。
[0410]
esi-hrms:c
59h97o14
[m+h
+
]的计算值1029.6873;测定值1029.6890。
[0411]
根据上述方法制得了下列通式(iv)的化合物:
[0412]
a2)3-甲基戊二酸1-(1,3-双(棕榈酰氧基)丙-2-基)5-(1-氯乙基)酯(xxviii)
[0413][0414]
将酸-tg iii(120mg,0.172mmol)、n,n-二甲基甲酰胺(dmf,1滴)和socl2(125μl,1.72mmol)的混合物回流加热1.25小时,然后冷却至室温。减压浓缩该反应体系,然后从甲苯中共蒸发3次(各3ml),减压干燥。将得到的酰氯再溶于ch2cl2(1.5ml),在0℃滴加到在ch2cl2(0.5ml)中的无水zncl2(23.5mg,0.172mmol),在0℃搅拌5分钟。加入三聚乙醛(45.6μl,0.344mmol),将该混合物在0℃搅拌10分钟,在室温搅拌1小时。用ch2cl2(20ml)稀释该反应体系,用水和盐水(各20ml)洗涤有机相,干燥(mgso4),减压浓缩,得到粗制产物。通过硅胶色谱法纯化(5%-15%乙酸乙酯/己烷),得到1-氯乙基酯xxviii(14.9mg,11%),为黄色油状物。
[0415]
b2)3-甲基戊二酸(e)-1-(1,3-双(棕榈酰氧基)丙-2-基)5-(1-((6-(4-羟基-6-甲氧基-7-甲基-3-氧代-1,3-二氢异苯并呋喃-5-基)-4-甲基己-4-烯酰基)氧基)乙基)酯(11)
[0416][0417]1h nmr(401mhz,cdcl3)δ7.68(s,1h),6.80(q,j=5.4hz,1h),5.29
–
5.21(m,2h),5.20(s,2h),4.32
–
4.26(m,2h),4.132/4.127(各自dd,j=11.9,6.0hz,2h),3.76(s,3h),3.38(d,j=6.9hz,2h),2.48
–
2.35(m,5h),2.32
–
2.18(m,8h),2.15(s,3h),1.79(s,3h),1.64
–
1.54(m,4h),1.41(d,j=5.4hz,3h),1.36
–
1.18(m,48h),1.01(d,j=6.4hz,3h),0.88(t,j=6.9hz,6h)。注意:双信号反映出存在非对映异构体混合物。
[0418]
esi-hrms:c
60h99o14
na[m+na
+
]的计算值1065.6849;测定值1065.6879。
[0419]
实施例9.大鼠中的淋巴转运研究
[0420]
为了评价本发明的化合物在大鼠中的淋巴转运,给大鼠肠系膜淋巴管插套管以允许连续采集肠系膜淋巴。然后将包含目标化合物的脂质制剂施用于动物,采集淋巴,随后对淋巴中的药物浓度定量。
[0421]
如先前所述制备了本发明的化合物或对照化合物的基于脂质的制剂(trevaskis,n.l.等人,pharmaceutical research,2005,22(11),1863-1870)。简言之,将约2mg的化合物(化合物19:1.5mg,且化合物6和7:0.05mg)、40mg油酸和25mg吐温80在玻璃小瓶中混合,直到平衡(适度加热(低于50℃)可以短期应用)。随后将由5.6ml(磷酸盐缓冲盐水(pbs,ph 7.4)组成的水相加到脂质相中,通过在室温下用超声处理机超声处理乳化该制剂2分钟,所述超声处理机安装有以240μm振幅和20khz频率运行的3.2毫米微探头尖。使用hplc-ms-ms验证全部制剂中的化合物浓度。
[0422]
选择雄性sprague-dawley(sd)大鼠用于淋巴转运研究,其中药用物质为麦考酚酸
(mpa)、舍曲林(ser)或丁丙诺啡(bup)。选择雌性sd大鼠用于其中睾酮为药用物质的研究。这是消除雄性大鼠中相对高和可变水平的内源性睾酮,它干扰外源性施用的睾酮的定量。将大鼠(220-320g)维持以标准膳食并且禁食过夜,可以自由引水,然后进行实验。在37℃将麻醉的大鼠置于加热垫上,如先前所述(edwards等人advanced drug delivery reviews 2001,50(1),45-60),将套管插入十二指肠(用于制剂施用和再水化)、肠系膜淋巴管(用于淋巴采集)和颈动脉(用于采血)。术后,将大鼠通过以2.8毫升/小时十二指肠输注生理盐水再水化0.5小时。将脂质制剂以2.8毫升/小时输注入十二指肠2小时,此后,将输注改变为2.8毫升/小时生理盐水,用于其余的实验。将淋巴连续采集直到8小时,进入预先称重的包含10微升的1,000iu/ml肝素的埃彭道夫管。每小时变换1次采集管,通过重量分析法测定淋巴流量。将每小时的淋巴样品的等分试样储存在-80℃,然后测定。
[0423]
将淋巴中的药物浓度表示为总药物且其包括游离药物以及与不同甘油酯结合的药物。通过水解淋巴(以从任意再酯化的甘油酯中释放药物)测定,然后评价游离药物。
[0424]
根据收集的淋巴体积和淋巴中测定的浓度的乘积,计算在每小时收集期间化合物向淋巴中的转运。
[0425]
如图1和表4所示,具有乙缩醛自消除基团(asi)的化合物12、具有三甲基-锁(tml)自消除基团的化合物15和具有(4-碳)翻转酯自消除基团(fsi-4)的化合物17的淋巴转运分别为(所施用剂量)的1.9%、3.2%和5.2%。这低于直链对应物睾酮琥珀酸-tg(化合物21,13.4%),其中化合物12降低至目前市售的睾酮前药十一烷酸睾酮(tu)的水平。含有自消除基团的前药的淋巴转运的减少可能是由于单酸甘油酯形式的前药在胃肠道中的稳定性差(如以下实施例11中所述和图9中所示),或可能是由于单酸甘油酯形式在肠细胞中的再酯化效率降低。化合物21、化合物12和化合物17的单酸甘油酯形式的稳定性次序(即化合物21-单酸甘油酯》化合物17-单酸甘油酯》化合物12-单酸甘油酯)与这三种前药的淋巴转运的排序一致(即化合物21》化合物17》化合物12)。
[0426]
在甘油酯单元的α碳或β碳位上包含甲基增加了前药的单酸甘油酯中间体的稳定性。例如,为了解决化合物12的稳定性问题,在化合物中包含甲基保护基团以形成化合物13和14(该化合物以化合物13和14的混合物形式使用)。正如从图1和表4中显见的,与化合物12相比,包含甲基保护基团显著增强了化合物13和14的淋巴转运。这与gi消化条件下增加的稳定性一致(参见图9)。化合物19(包含带有甲基支链的5-碳翻转酯自消除基团[fsi-5])的淋巴转运为(所施用剂量的)9.6%。这也高于类似的化合物,即化合物17(包含fsi-4自消除基团)但缺乏甲基。
[0427]
表4.十二指肠内输注给麻醉的肠系膜淋巴管插管的大鼠后总化合物的淋巴转运(所施用剂量的%)(当n≥3时,数据表示为平均值
±
sem,或当n=2时,数据表示为平均值
±
范围)。
[0428][0429]
*睾酮在淋巴样品中的浓度低于定量极限。
[0430]
正如图4和表5中所示,具有tml自消除基团的化合物3的淋巴转运为(所施用剂量的)21.5%。与之形成鲜明对比的是,母体药物ser.hcl施用后母体药物舍曲林(ser)的淋巴转运仅为(所施用剂量的)0.05%。
[0431]
表5.十二指肠内输注给麻醉的肠系膜淋巴管插管的大鼠后总化合物的淋巴转运(所施用剂量的%)(当n≥3时,数据表示为平均值
±
sem,或当n=1时,数据表示为单个值)。
[0432][0433]
正如图6和表4中所示,具有tml自消除基团的化合物6和具有(5-碳)翻转酯自消除团(fsi-5)的化合物7的淋巴转运为(所施用剂量的)10.8%和24.5%。相反,bup施用后母体药物丁丙诺啡(bup)的淋巴转运仅为(所施用剂量的)0.01%。
[0434]
表6.十二指肠内输注给麻醉的肠系膜淋巴管插管的大鼠后总化合物的淋巴转运(所施用剂量的%)(当n≥3时,数据表示为平均值
±
sem,或当n=2时,数据表示为平均值
±
范围)。
[0435]
[0436]
正如图8和表7中所示,具有乙缩醛自消除(asi)基团的化合物10和具有甲基乙缩醛自消除(masi)基团的化合物11的淋巴转运为(所施用剂量的)0.35%和1.22%。与之形成鲜明对比的是,母体药物mpa施用后母体药物麦考酚酸(mpa)的淋巴转运仅为(所施用剂量的)0.17%。
[0437]
表7.十二指肠内输注给麻醉的肠系膜淋巴管插管的大鼠后总化合物的淋巴转运(所施用剂量的%)(当n≥3时,数据表示为平均值
±
sem,或当n=2时,数据表示为平均值
±
范围)。
[0438][0439]
实施例10.大鼠中的药物动力学(pk)研究
[0440]
为了评价本发明化合物的口服生物利用度,使用以下方法进行药物动力学研究。施用前一天,麻醉雌性(用于睾酮相关研究)或雄性(用于ser和bup相关研究)sprague-dawley大鼠(220-320g),并且给颈动脉插管。然后允许大鼠恢复意识并禁食过夜,然后开始实验,可随意饮水。第二天早上,通过经口灌胃施用含有母体化合物或前药的制剂,并且在施用后-5分钟至24小时从颈动脉插管收集血样,并以5000rpm离心5分钟以分离血浆。在血样采集期间,大鼠可自由饮水,但在给药后再保持禁食8小时。在通过hplc-ms-ms分析之前,将血浆样品储存在-80℃。在这种情况下,分析样品的游离药物(即非甘油酯结合的药物),并且在分析之前不水解(如淋巴样品的情况)。因此该数据反映了被运输到淋巴中且然后从体循环中的再酯化的药物-甘油酯复合物释放的药物。
[0441]
如先前所述,在药用物质睾酮和甘油酯单元之间使用短连接基的甘油三酯前药(例如琥珀酸,化合物21)受到体循环中药物不良释放的限制(参见scriba,g.k.e.,arch.pharm.(weinheim)。1995,328,(3),271-276;以及scriba,g.k.e.等人,j.pharm.pharmacol.1995,47,(11),945-948))。如下图2和表8所示,将自消除基团添加到连接基上增加了药用物质的系统暴露,从而避免了活性剂的首过代谢,并增加了其口服生物利用度,即使当与先前弃用的琥珀酸连接基组合时也是如此。
[0442]
图2示例了对有意识的颈动脉插套管的雌性sd大鼠经口管饲睾酮后剂量归一化睾酮血浆浓度。制剂含有1mg tu或tst,或约2mg本发明化合物,含有分散于40mg油酸、25mg吐温80和2ml pbs中的睾酮。将剂量归一化2mg/kg相当剂量的睾酮。数据显示为平均值
±
sem。嵌入式图是棒形图形式的剂量归一化血浆auc
0-24h
(nmol
×
h/l)睾酮的图。
[0443]
表8显示经口施用化合物12-20或21后,母体睾酮的药物动力学参数。在短链连接基中不含甲基的所有情况下(化合物12、15-18或20),睾酮的系统暴露大于化合物21的系统暴露(约~1.4
–
18倍增加)或商业产品tu的系统暴露(~4
–
54倍增加)。尽管化合物12和化合物17的淋巴转运相对低(图1和表4),但经口施用两种前药后,母体睾酮的系统暴露仍然高很多。这表明,所述的自消除基团促进前药在体循环中转化为母体药物。
[0444]
表8.给有意识的颈动脉插套管的sd雌性大鼠经口施用所述化合物后的药物动力学参数(将剂量归一化为2mg/kg相当的睾酮剂量且将数据表示为平均值
±
sem)。
[0445][0446]
前药的实用性可通过增加前药的淋巴运输得到进一步增强。将甲基包含在短链连接基任一末端上的酯的α碳或β碳位上能够增加前药的单酸甘油酯中间体的稳定性,从而增加淋巴转运。化合物12对比化合物13/14的体外消化和淋巴转运结果(参见图9、图1和表4)支持该启示,并显示在模拟肠道条件下化合物13/14显著增强的稳定性。与化合物13/14的淋巴运输增加以及自消除基团促进睾酮从前药系统释放的潜力一致,单独施用化合物13/14或化合物13后睾酮的系统暴露比化合物12高~13倍,比tu高95-97倍(表8)。此外,在fsi前药的情况下,将甲基包含在短链连接基上还提高了睾酮的生物利用度。因此,化合物19施用后睾酮的系统暴露比化合物18高1.9倍,比tu高105倍(表8)。
[0447]
图5示例对有意识的颈动脉插套管的雄性sd大鼠经口管饲制剂后剂量归一化ser血浆浓度。在ser母体药物对照组中,制剂含有溶于2ml水中的0.7mg ser.hcl。前药制剂包含2mg本发明化合物,含有分散在40mg油酸、25mg吐温80和2ml pbs中的ser。将剂量归一化为ser的2mg/kg相当剂量。数据显示为平均值
±
sem。嵌入式图是棒形图形式的ser的剂量归一化血浆auc
0-24h
(nmol
×
h/l)的图。
[0448]
表9中显示了ser.hcl、化合物1、2和3施用后ser的药物动力学参数。在所有情况下,施用所述前药后ser的系统暴露大于ser.hcl的系统暴露(2-3倍增加)。这表明,所述的前药是通过淋巴转运的(如图4和表5中的化合物3所例证的),并且所述的自消除基团促进了前药在体循环中转化为母体药物。
[0449]
表9.给有意识的颈动脉插套管的sd雄性大鼠经口施用ser相关化合物后的药物动力学参数(将剂量归一化为2mg/kg相当的ser剂量且将数据表示为平均值
±
sem)。
[0450][0451]
图7示例对有意识的颈动脉插套管的雄性sd大鼠经口管饲制剂后剂量归一化bup血浆浓度。在bup母体药物对照组中,制剂含有溶于2ml 0.1%乙酸水溶液中的0.02mg bup。前药制剂包含0.05mg本发明化合物,含有分散在40mg油酸、25mg吐温80和2ml pbs中的bup。将剂量归一化为bup的0.06mg/kg相当的剂量。数据显示为平均值
±
sem。嵌入式图是棒形图形式的bup的剂量归一化血浆auc
0-6h
(nmol
×
h/l)的图。
[0452]
表10中显示了bup、化合物5、6和7施用后bup的药物动力学参数。在所有情况下,施用本发明的化合物后bup的系统暴露大于bup的系统暴露(7-14倍增加)。这表明,所述的前药是通过淋巴转运的(如图6和表6中化合物6和7所例证的),并且所述的自消除基团促进了前药在体循环中转化为母体药物。
[0453]
表10.给有意识的颈动脉插套管的sd雄性大鼠经口施用bup相关化合物后的药物动力学参数(将剂量归一化为0.06mg/kg相当的bup剂量且将数据表示为平均值
±
sem)。
[0454][0455]
*使用截短的aucs(auc
0-6h
)而非auc
0-24h
,这是因为在施用母体药物后bup血浆浓度与时间关系图中存在第二个峰。第二个峰最有可能反映bup的肠肝再循环。
[0456]
实施例11.猪胰酶脂酶对化合物的体外水解
[0457]
通过与猪胰脂肪酶一起温育进行睾酮前药的体外水解。简言之,在水解实验之前,将1g猪胰酶制剂分散在5ml脂解缓冲液中,制备胰脂肪酶溶液。充分混合悬浮液并在5℃以3500rpm离心15分钟以提供上清液。用0.474克tris-马来酸盐(2mm)、0.206克cacl2.h2o(1.4mm)和8.775克nacl(150mm)制备了1000毫升量的脂解缓冲液,用naoh调节至ph 6.5。为了评价前药在肠中水解的可能性,将20微升前药溶液(1mg/ml溶于乙腈)、900微升模拟肠胶束溶液[用0.783克natdc(3mm)和0.291克磷脂酰胆碱(0.75mm)于500毫升脂解缓冲液中制备]和100微升酶溶液在37℃下温育。温育后0、5、10、15、30、60、90、120和180分钟时取出20微升温育溶液样品,并加到180微升acn中以停止脂解。涡旋混合物并在5000rpm下离心5分钟以在分析之前沉淀蛋白质。通过hplc-ms分析上清液中的残余化合物浓度,并分析化合物
水解的潜在产物。
[0458]
与消化酶一起温育时,非常迅速地形成前药的单酸甘油酯形式。因此,用初始消化过程产生的单酸甘油酯形式的稳定性更好地评价模拟肠道条件下的稳定性。单酸甘油酯形式在进入淋巴系统之前必须保持完整以在肠细胞中被吸收和再酯化。图9中,将与新鲜制备的猪胰脂肪酶体外温育过程中化合物12、化合物15-17和化合物20(每组n=2-3)的单酸甘油酯形式的稳定性曲线与含有非自消除基团的化合物21的稳定性曲线(n=3)比较。数据显示,包含所述的乙缩醛(asi)、翻转酯(fsi)、羧基(甲基乙缩醛)(cmsi)或对羟基苄基羰基(phb)自消除基团导致前药的mg形式的管腔稳定性显著降低。然而,因为化合物15的单酸甘油酯形式与化合物21的单酸甘油酯形式类似稳定,所以三甲基锁自消除基团似乎对管腔稳定性没有影响。
[0459]
图9还提供了甲基取代改善含有乙缩醛自消除连接基的睾酮前药的腔稳定性的能力的证据(其中乙缩醛基团通常降低管腔稳定性)。如该图中所示,化合物12的单酸甘油酯形式在体外脂解试验中是不稳定的。相反,α和β甲基取代的化合物13/14的混合物更稳定得多。甲基取代的asi前药的增强稳定性最有可能是体内淋巴转运显著增加以及经口施用后体循环中睾酮暴露睾酮增加的原因。
[0460]
实施例12.在补充了脂蛋白脂酶的淋巴中mpa从前药中体外释放
[0461]
为了探究淋巴系统中从tg前药释放游离mpa(mpa的活性位点在淋巴系统中富含的淋巴细胞中),将mpa前药与补充了脂蛋白脂肪酶(lpl,200单位/毫升)的大鼠淋巴一起温育。lpl是在正常生理条件下水解脂蛋白结合的tg所需的关键酶,因此预期它是血浆中重新酯化的药物-tg构建体的脂解的关键贡献者,主要是通过释放tg-模拟物的sn-1和sn-3位的fa来进行,然后通过酯酶水解从2'位置释放药物。lpl在生理条件下与淋巴细胞或淋巴系统/血管内皮细胞连接。目前在体外研究中,因此大鼠淋巴补充了lpl以更好地反映体内情况。为了开始水解,将10微升lpl溶液(10,000单位/毫升)加到10微升前药溶液(1mg/ml溶于乙腈中)和500微升空白sprague dawley大鼠淋巴的混合物中。将该溶液在37℃温育。温育后0、5、10、15、30、60、90、120和180分钟取出温育溶液的样品(20微升),并加到980微升9:1(v/v)acn-水中以终止脂解。涡旋混合物并在4500g离心5分钟以沉淀蛋白质,然后分析。通过hplc-ms/ms分析上清液的mpa浓度。
[0462]
如图10(每种前药n=1)中所示,在与lpl补充的大鼠淋巴一起温育时,从化合物10和化合物11中释放药理活性的mpa非常迅速。母体mpa从这两种含有si基团的前药中的释放速率远高于从不含si基团的前药mpa-tg中的释放速率。数据显示,包含乙缩醛(asi)或甲基乙缩醛(masi)自消除基团导致母体化合物的释放显著增强,因此提供了将治疗剂靶向递送至淋巴系统中的活性位点的机会。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1