DNA甲基化测序文库的制备方法和甲基化检测方法与流程
dna甲基化测序文库的制备方法和甲基化检测方法
技术领域
1.本技术涉及基因检测技术领域,具体涉及一种dna甲基化测序文库的制备方法和甲基化检测方法。
背景技术:
2.生物体内的dna甲基化的水平通常与诱导或抑制基因的表达有关。如果dna的甲基化发生异常,则可能与肿瘤的发生、发展有关,因此,通过获得特定基因的甲基化水平对于肿瘤早筛有重要的价值。
3.目前通过高通量测序平台来对特定基因进行测序,这首先需要根据特定基因来建立dna甲基化测序文库。现有的dna甲基化测序文库的建库方法包括:基于亚硫酸盐转化法的单链建库法和基于酶学转化法的双链建库法。
4.如图1所示,基于亚硫酸盐转化法的单链建库法包括如下步骤:将目标双链dna变性为单链dna;通过化学反应改变单链dna中胞嘧啶上的甲基,以产生受保护基团保护的胞嘧啶;利用转化试剂亚硫酸盐将未甲基化的胞嘧啶转化为尿嘧啶,然后建立用于供测序平台测序的dna甲基化文库。该方法的性能比较稳定,但是亚硫酸盐处理对dna的损伤较大,特别是针对末端缺失的双链dna,会造成dna断链、碱基丢失(例如形成无碱基位点)、甲基化信息失真等现象。
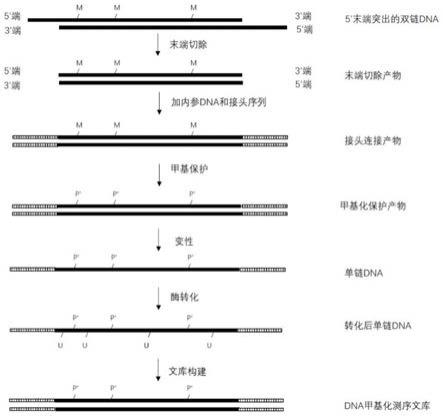
5.如图2所示,基于酶学转化法的双链建库法包括如下步骤:将目标双链dna进行3’末端修复,并直接加接头序列,接头序列里的胞嘧啶(c)为全部甲基化(如图2中m所示)的胞嘧啶,在双链状态下通过化学反应改变单链dna中胞嘧啶上的甲基,以产生受保护基团保护的胞嘧啶,然后变性为单链dna;采用转化酶将未甲基化的胞嘧啶转化为尿嘧啶(u),然后通过pcr扩增来建立用于供测序平台测序的dna甲基化文库。因为接头序列在最开始已经加上了,因此,转化后直接通过pcr扩增来构建文库。该方法采用未甲基化的dntp对双链dna的缺失末端进行修复,会造成甲基化信息的丢失,特别是对于血液样本,可能造成50%的甲基化信息丢失。另外,由于测序结果是针对末端修复后的双链dna,难以了解原始的双链dna到底有多少个碱基缺失以及多少个碱基被修补,导致这些失真的甲基化信息无法在后续生物信息学分析中修正。进一步地,测序出来的dna序列中的碱基数目多于原始的dna序列的碱基数目,这会造成数据污染。
技术实现要素:
6.本技术提供一种dna甲基化测序文库的制备方法和甲基化检测方法,其采用的试剂性质温和,对dna结构的损伤小,不会造成dna断链、碱基丢失等现象,也不会产生由于采用未甲基化的dntp进行末端修复而导致的甲基化信息失真和数据污染等现象。
7.为了克服上述缺点,本技术采用了以下技术方案:
8.[dna甲基化测序文库的制备方法]
[0009]
本技术提供一种dna甲基化测序文库的制备方法,其包括如下步骤:
[0010]
(1)、获得末端突出的双链dna;末端突出的双链dna的突出末端可以位于该双链dna的5’末端,也可以位于3’末端;
[0011]
(2)、采用核酸外切酶切除末端突出的双链dna的突出末端(jagged ends),以形成具有平末端(blunt ends)的双链dna,即末端切除产物;该末端突出的双链dna的用量可以在25-400ng的范围内,还可以在30-400ng的范围内;本技术的核酸外切酶7号能够切除双链dna的突出末端,包括突出的5’末端和3’末端;
[0012]
(3)、将平末端的双链dna与内参dna混合,得到dna混合样品;本步骤中,无需在平末端的双链dna的两个末端添加腺嘌呤重复序列(poly-a),在加入内参dna之后,可以直接在双链dna的末端连接测序用的接头序列;
[0013]
(4)、在dna混合样品中所有双链dna的两个末端添加接头序列,得到接头连接产物;
[0014]
(5)、采用甲基化保护试剂将接头连接产物中的双链dna的甲基化胞嘧啶氧化为受保护胞嘧啶,得到甲基化保护产物;
[0015]
(6)、将甲基化保护产物中的双链dna变性为单链dna;
[0016]
(7)、采用转化试剂将单链dna中未甲基化的胞嘧啶转化为尿嘧啶,得到转化后单链dna;
[0017]
(8)、对转化后单链dna进行pcr扩增,得到dna甲基化测序文库。
[0018]
其中,在一些实施例中,在步骤(1)中,末端突出的双链dna可以为血液中游离的cfdna。该末端突出的双链dna可以为5’末端突出的双链dna(或称为3’末端缺失的双链dna,以下同理),也可以为3’末端突出的双链dna(或称为5’末端缺失的双链dna,以下同理)。
[0019]
在一些实施例中,cfdna在血液中可以主要以缠绕核小体的形式存在,因为在该种情况下没有缠绕在核小体上的cfdna会很快被降解。一个核小体上缠绕的cfdna大约170bp,因此,cfdna的主要片段在170bp存在一个主峰(相当于一个核小体),然后在340bp有一个小峰(相当于两个核小体),以此类推。
[0020]
在另一些实施例中,cfdna的长度也可以位于170bp至210bp之间(包括本数)。然而,在其它一些实施例中,cfdna的长度也可以为180bp、185bp、190bp、195bp、200bp等。
[0021]
在其它的一些实施例中,cfdna的长度可以为170bp至210bp之间的任意一个数值的整数倍(根据所缠绕的核小体的个数确定),例如为340bp、360bp、380bp、400bp、420bp等。
[0022]
在一些实施例中,在步骤(1)中,末端突出的双链dna也可以为ctdna。如果将血液中末端突出的dna视为总cfdna,那么ctdna可以是cfdna中来自于肿瘤细胞的部分,即其来自于组织细胞的非正常凋亡过程,因此,同一来源或不同来源的单个不同的ctdna的片段大小不同,其片段的均一性小于组织细胞的正常凋亡过程形成的cfdna。因此,本技术文件中的cfdna可以定义为正常组织的cfdna(即不包括非正常凋亡所产生的ctdna),而将ctdna定义为特指肿瘤细胞的非正常凋亡所产生的血液中游离的末端缺失的双链dna。另外,该末端突出的双链dna可以为5’末端突出的双链dna(或称为3’末端缺失的双链dna),也可以为3’末端突出的双链dna(或称为5’末端缺失的双链dna)。
[0023]
在一些实施例中,ctdna的长度可以位于60bp至350bp之间(包括本数)。例如,ctdna的长度也可以为60bp、70bp、75bp、80bp、100bp、120bp、150bp、160bp、180bp、200bp、210bp、220bp、230bp、250bp、270bp、280bp、300bp、320bp、350bp等。
[0024]
在一些实施例的步骤(1)中,末端突出的双链dna也可以为具有突出末端的基因组dna(genomic dna,gdna),这是因为基因组dna处于游离状态时,其3’末端或5’末端可能会被某些酶消化或者受到某些化学试剂的作用等因素而产生末端缺失,从而与该缺失末端并列的另一末端呈现突出状态。在构建测序文库时,该基因组dna因为片段较长,需要使用打断仪打断为240bp至290bp之间(包括本数),例如,打断至250bp、260bp、270bp、280bp等。在一些实施例中,其端点值可以参照上述的cfdna或ctdna的取值。另外,该末端突出的基因组双链dna可以为5’末端突出的双链dna(或称为3’末端缺失的双链dna),也可以为3’末端突出的双链dna(或称为5’末端缺失的双链dna)。
[0025]
在步骤(1)中,末端突出的双链dna的每条链的突出末端可以具有1至80个碱基中的任意一个整数值,也可以具有5至70个碱基中的任意一个整数值,也可以为10至50个碱基中的任意一个整数值,可以进一步为20至30个碱基中的任意一个整数值。在其它一些实施例中,上述的双链dna的每条链的5’末端或3’末端的碱基突出数可以为2、3、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、25、35、45、55、65、75等。
[0026]
在步骤(1)中,对双链dna的突出的5’端和/或3’端进行了切除,因此,本技术的用于测序的双链dna的总数据量相对于采用dntps进行3’末端修复的基于酶学转化法的双链建库法而言有了降低,因此,本技术的dna的起始量需要比基于酶学转化法的双链建库法所采用的dna的起始量要高,以便对因切除而损失的那部分数据量进行补偿,从而在后继的甲基化水平的计算时将其计算在内。与基于酶学转化法的双链建库法相比,本技术的双链dna的起始量的调高的百分比至少等于、并且甚至高于该双链dna的末端切除率或平均末端切除率。
[0027]
在一些实施例中,本技术中的末端切除率x被定义为双链dna样品中单个双链dna分子的5’或3’末端切除的最大碱基数占该双链dna分子的总的碱基数的百分比。为了更精确地进行计算,末端切除率的分子选取末端被切除的最大碱基数,而不是双链dna样品中末端被切除的最小碱基数,或者平均碱基数。示例性地,在双链dna样品中,如果同一来源的双链dna有m个,每个双链dna的原始长度为l,部分双链dna的5’末端或3’末端的突出长度为1个至n个(n《l),其中n的取值为2至80中的任意一个数值,l的取值为170至210bp之间,或者为170至210bp之间的任意一个数的整数倍。在该种情况下,n即为该双链dna样品的5’末端或3’末端切除的最大碱基数,作为末端切除率的分子,并且l的取值作为分母,那么末端切除率x等于n/l
×
100%。
[0028]
在一些实施例中,本技术中的平均末端切除率x’被定义为双链dna样品中所有双链dna分子的5’或3’末端切除的平均碱基数占该双链dna分子的总的碱基数的百分比。示例性地,在双链dna样品中,如果同一来源的双链dna有m个,每个双链dna的原始长度为l,部分双链dna的5’末端或3’末端的突出长度为1个至n个(即这些双链dna分子的末端缺失程度存在一定差异),其中n的取值为2至80中的任意一个数值,l的取值为170至210bp之间,或者为170至210bp之间的任意一个数的整数倍。在该种情况下,n’被确定为该双链dna样品中所有双链dna分子的5’末端或3’末端切除的平均碱基数,作为平均末端切除率的分子,并且l的取值作为分母,那么平均末端切除率x’等于n’/l
×
100%。n’可以经过换算或估算得到。其中一种估算法为n’=(1+n)/2。
[0029]
进一步地,如果未进行末端切除的建库法(如基于亚硫酸盐转化法的单链建库法,
或者基于酶学转化法的双链建库法)所采用的双链dna的建库起始量为y,那么本技术所采用的双链dna的建库起始量y’至少为y
×
(1+x),甚至可以为y
×
(1+nx),n可以为2至5倍中的任意一个数值,也可以为3至4倍中的任意一个数值等。或者,本技术所采用的双链dna的建库起始量y’至少为y
×
(1+x’),甚至可以为y
×
(1+nx’),n可以为2至5倍中的任意一个数值,也可以为3至4倍中的任意一个数值等。
[0030]
示例性地,如果基于亚硫酸盐转化法的单链建库法的双链dna的起始量y为20ng,则本技术所采用的双链dna的建库起始量y’为20ng
×
(1+nx),其中n的值为1倍至5倍中的任意一个数值,x指代末端切除率。平均末端切除率x’的情况同理。
[0031]
在步骤(2)中,末端切除过程可以利用末端切除体系进行。末端切除体系可以采用核酸外切酶7号(exonuclease vii)等进行末端切除。核酸外切酶能够依次降解双链dna的突出末端。末端切除的反应条件为:在37℃的温度条件下反应30min,然后在95℃的温度条件下反应10min,最后在4℃的温度条件下保存。
[0032]
在步骤(3)中,内参dna包括λdna和puc19 dna。
[0033]
在步骤(5)中,甲基化保护试剂包括tet2蛋白和氧化增强剂。tet2蛋白用于将甲基化胞嘧啶中的甲基经过氧化反应转换为保护基团(例如,甲酰基或羧基)。受到保护基团保护的胞嘧啶能够对抗胞嘧啶脱氨酶的脱氨作用,从而不被转换为尿嘧啶。未甲基化的胞嘧啶不存在甲基,不会连接上保护基团,从而不能够对抗胞嘧啶脱氨酶的脱氨作用,后继会被转化为尿嘧啶。
[0034]
在步骤(5)中,甲基化胞嘧啶为5-甲基胞嘧啶或5-羟甲基胞嘧啶;
[0035]
在步骤(5)中,受保护胞嘧啶为5-甲酰基胞嘧啶或5-羧基胞嘧啶。
[0036]
在步骤(6)中,变性的温度为50
±
5℃,添加的变性剂为强碱,例如为naoh。naoh的浓度为0.1mol/l。针对16μl的甲基化保护产物,加入的naoh的量为4μl。
[0037]
在步骤(7)中,转化试剂包括apobec蛋白。apobec蛋白具有胞嘧啶脱氨酶的活性,能够使得未甲基化的胞嘧啶脱氨,从而转化为尿嘧啶。
[0038]
在步骤(8)中,pcr扩增过程利用pcr扩增体系进行。pcr扩增体系包括20μl的转化后单链dna、5μl的em-seq index primer plate和25μl的nebnext q5u master mix。所使用的试剂盒为nebnext enzymatic methyl-seg kit。pcr的反应条件为:先在98℃时运行30s;然后在98℃时持续10s,62℃时持续30s,64℃时持续60s,一共持续9个循环;接着在65℃时持续5min,最后在4℃时保存。
[0039]
[dna甲基化测序文库]
[0040]
本技术提供了一种dna甲基化测序文库,该dna甲基化测序文库由上述的制备方法构建而成。该dna甲基化测序文库的每个dna分子的两个平头端带有测序用的接头序列,能够被测序仪读取,从而能够测得该dna的甲基化水平。
[0041]
[dna甲基化水平的检测方法]
[0042]
本技术提供了一种dna甲基化水平的检测方法,其包括如下步骤:
[0043]
(1)、对5’末端或3’末端突出的双链dna按照上述的制备方法构建dna甲基化测序文库;
[0044]
(2)、采用测序仪对dna甲基化测序文库进行测序,得到测序结果;
[0045]
(3)、对测序结果进行分析,得到dna甲基化水平。
[0046]
本技术对测序结果的生物信息学分析过程与上述的基于亚硫酸盐转化法的单链建库法和基于酶学转化法的双链建库法相同。本技术对因为剪切末端所带来的数据量损失并非通过生物信息学过程的分析来进行补偿,而是通过一定程度上提高双链dna的建库起始量进行补偿。因为从建库到测序的过程中涉及“随机抽样”的过程,再加上上述的补偿过程,所以剪切末端所带来的数据量的损失对有效数据量而言基本不存在影响,那么也就对dna的甲基化测序结果而言基本不存在影响。
[0047]
由于采用了上述技术方案,本技术取得了如下的技术效果:
[0048]
首先,本技术采用核酸外切酶7号切除末端突出的双链dna中的5’突出末端或3’突出末端,使其成为平末端的双链dna,然后在两个末端加上内参dna和测序用的接头序列,并且以此来构建dna甲基化测序文库。由于本技术并未采用未甲基化的dntps进行末端修复,不会在末端引入新的甲基化水平,不会产生甲基化水平失真现象。并且,本技术通过提高双链dna的建库起始量来弥补由于末端切除部分碱基而造成的dna整体数据量降低的现象,因此,通过双链dna的末端切除和提高dna的建库起始量相结合的方法,再加上从建库到测序中“随机抽样”的过程的影响,本技术最终测得的dna甲基化水平非常接近于真实的甲基化水平,所产生的误差在商业化测序时可以忽略不计。
[0049]
另外,本技术在将未甲基化的胞嘧啶转化为尿嘧啶时采用反应条件温和的酶学转化法,而并未添加亚硫酸盐,因此,不会造成dna断链、碱基丢失等现象。
附图说明
[0050]
为了更清楚地说明本技术实施例中的技术方案,下面将对实施例描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本技术的一些实施例,对于本领域技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
[0051]
图1为基于亚硫酸盐转化的单链建库法的步骤示意图。
[0052]
图2为基于酶学转化的双链建库法的步骤示意图。
[0053]
图3为本技术的基于5’末端剪切和酶学转化的双链建库法的步骤示意图。
[0054]
图4为本技术的基于3’末端剪切和酶学转化的双链建库法的步骤示意图。
[0055]
图5为本技术与现有酶学转化甲基化建库的测序结果对比结果图。
[0056]
上述各图中的m表示甲基化胞嘧啶上的甲基。p
*
表示受保护胞嘧啶上的保护基团。上述各图只显示了双链dna中其中一条链的处理方法,另一条链作同样处理,故上述各图的部分区域仅仅显示其中一条链,并不代表另外一条链就不能作同样处理。在测序文库构建的过程中,尿嘧啶u会转换为胸腺嘧啶t,故最终的测序文库不再显示尿嘧啶。上述各图中的黑色区域表示dna的链,空白区域和/或花纹区域等表示额外添加的接头序列等。不同的花纹区域表示不同的序列。
具体实施方式
[0057]
下面将结合本技术实施例中的附图,对本技术实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本技术的一部分实施例,而不是全部的实施例。基于本技术中的实施例,本领域技术人员在没有作出创造性劳动前提下所获得的所有其他实
施例,都属于本技术保护的范围。
[0058]
以下分别对本技术进行详细说明。需说明的是,以下实施例的描述顺序不作为对实施例优选顺序的限定。
[0059]
除非另有说明,文中涉及的试剂与材料均可商购获得,或本领域技术人员可依据公知常识自行制备。除非另有说明,文中的技术术语的含义与公知常识或技术词典中的含义相同。
[0060]
如图3和图4所示,本技术提供了一种dna甲基化测序文库的制备方法,其包括如下步骤:
[0061]
(1)、获得末端突出的双链dna(又称为带有突出末端的双链dna);该末端突出的双链dna的突出末端可以位于该双链dna的5’末端,也可以位于3’末端;
[0062]
(2)、采用核酸外切酶切除末端突出的双链dna的5’突出末端和/或3’末端,以形成具有平末端的双链dna,即末端切除产物;
[0063]
(3)、将末端切除产物中平末端的双链dna与内参dna混合,得到dna混合样品;
[0064]
(4)、在dna混合样品中的所有双链dna的两个末端添加测序用的接头序列,得到接头连接产物;
[0065]
(5)、采用甲基化保护试剂将接头连接产物中的双链dna的甲基化胞嘧啶氧化为受保护胞嘧啶,得到甲基化保护产物;
[0066]
(6)、将甲基化保护产物中的双链dna变性为单链dna;
[0067]
(7)、采用转化试剂将单链dna中未甲基化的胞嘧啶转化为尿嘧啶,得到转化后单链dna;
[0068]
(8)、对转化后单链dna进行pcr扩增,得到dna甲基化测序文库。
[0069]
其中,在步骤(1)中,双链dna的获取方法可以采用常规的dna提取方法。
[0070]
在步骤(1)中,所获得的双链dna为5’和/或3’末端突出的双链dna。生物体的血液中往往会出现处于游离状态并且末端突出的双链dna,例如,由组织细胞经由正常的凋亡过程而产生的循环游离dna(circulating free dna,cfdna),或由肿瘤细胞经由非正常的凋亡过程而产生的循环肿瘤dna(circulating tumor dna,ctdna)等。
[0071]
正常的cfdna主要是通过细胞凋亡过程中产生的,长度呈均匀分布并且位于170至210bp之间,或者为170至210bp之间的任意一个自然数值的倍数,浓度一般小于或等于100ng/ml,平均浓度约为30ng/ml。
[0072]
ctdna则是在非正常凋亡过程中产生,其特异性地来自于肿瘤细胞,故能够用作对肿瘤细胞的超早期筛查。该ctdna的长度呈大小不同分布并且位于60bp至350bp之间(包括本数),浓度按肿瘤进展期不同一般为0.01至10ng/ml,平均浓度约为0.1ng/ml这些双链dna在进入血液中后,由于受到血液中多种酶和化学物质的作用,其末端往往存在1-80个碱基的缺失,从而呈现双链不平整的状态。
[0073]
本技术中,如果待测样本来自于5’和/或3’末端突出的基因组dna(genomic dna,gdna),则需要将该待测样本中的dna打断为240bp至290bp,以得到片段化的dna,然后再构建dna甲基化测序文库。如果待测样本来自于cfdna,由于cfdna本身的片段较短,故无需打断过程。组织细胞提取的dna也会存在不平整的粘性末端,但是由于组织dna一般的片段的长度较长,实验中需要通过超声波机械打断成短片段,而超声打断的“切口”一般较为整齐,
所以相对来说不平整末端的影响并不那么大。
[0074]
在步骤(2)中,该末端突出的双链dna的用量可以为25-400ng,也可以为30-240ng,还可以为50-120ng。该用量根据末端切除的碱基数进行确定若末端切除的碱基数较多,则双链dna的用量较高;若末端切除的碱基数较少,则双链dna的用量较低。但总的而言,本技术的双链dna的用量应高于未进行末端切除的双链dna的用量。未进行末端切除的双链dna所使用的建库法为基于亚硫酸盐转化法的单链建库法或基于酶学转化法的双链建库法。
[0075]
本技术通过提高双链dna的起始量(即用来构建文库的dna量)来弥补整体数据量的降低。研究表明,与没有外切粘性末端的方法所得的数据量相比,本技术通过外切去除粘性末端造成的数据量损失在10%以下。在pcr扩增后,并非所有的文库都上机测序,而在上机测序时,测序仪的测序接头连接也是仅有少量分子结合,大部分被遗弃,故从建库到测序过程中实际涉及多处“随机抽样”过程,很难对“数据损失”有准确定量的描述。因此,对切除粘性末端的方法与不切除粘性末端的建库法之间的数据量的比较是根据有效数据量进行的,而并非根据绝对数据量进行的。经过实验证明,本技术的方法由于没有引入错误的甲基化信号,而不切除粘性末端的现有酶学建库法由于采用了末端修复而引入了错误的甲基化信号,那么针对同一来源的样本,本技术在保证所测得的甲基化水平与真实的甲基化水平基本一致的前提下,虽然因为经过剪切末端而使得绝对数据量有了下降,但是有效数据量却比现有neb的酶学建库法要高。
[0076]
在步骤(3)中,所述内参dna包括λdna和puc19 dna。
[0077]
在步骤(5)中,甲基化保护试剂包括tet2蛋白和氧化增强剂。
[0078]
在步骤(5)中,甲基化胞嘧啶为5-甲基胞嘧啶或5-羟甲基胞嘧啶,受保护胞嘧啶为5-甲酰胞嘧啶或5-羧基胞嘧啶。
[0079]
在步骤(6)中,变性的温度为50
±
5℃,添加的变性剂为强碱液,例如为naoh的水溶液,其浓度为0.1mol/l。
[0080]
在步骤(7)中,转化试剂包括apobec蛋白。
[0081]
在步骤(8)中,pcr扩增在pcr扩增体系中进行,pcr扩增体系包括20μl的转化后单链dna、5μl的em-seq index primer plate和25μl的nebnext q5umaster mix;pcr的反应条件为:先在98℃时运行30s;然后在98℃时持续10s,62℃时持续30s,64℃时持续60s,一共持续9个循环;接着在65℃时持续5min,最后在4℃时保存。
[0082]
本技术还提供了一种dna甲基化测序文库,该dna甲基化测序文库由上述任意一种制备方法或其组合构建而成。
[0083]
本技术还提供了一种dna甲基化水平的检测方法,其包括如下步骤:
[0084]
(1)、对带有不平整末端(又称粘性末端)的双链dna按照上述的任意一种制备方法或其组合构建dna甲基化测序文库;
[0085]
(2)、采用测序仪对dna甲基化测序文库进行测序,得到测序结果;本技术的测序采用illumina公司的测序仪和测序方法;
[0086]
(3)、对测序结果进行分析,得到dna甲基化水平。
[0087]
在本技术中,dna样品没有经过重亚硫酸盐处理,而是经过较温和的酶处理,dna的结构不会受损伤,使得测得的甲基化水平更加接近真实值。
[0088]
本技术的dna甲基化测序文库能够适用于illumina高通量测序平台,所得到的dna
甲基化水平非常精确,能够绘制单碱基分辨率的细胞游离dna的甲基化图谱。
[0089]
本技术采用末端剪切、酶学转化和双链建库法相结合的dna测序文库制备方法,其采用核酸外切酶切除双链dna中的突出的不平整末端,然后加入内参dna和测序用的接头序列,而无需在两个末端加上腺嘌呤重复序列,之后,变性后采用酶学转化法将单链dna中未甲基化的胞嘧啶转化为尿嘧啶,通过pcr进行扩增,以得到dna甲基化测序文库。因为本技术并未采用未甲基化的dntp进行末端修复,不会在末端引入新的甲基化水平。因此,本技术能够避免采用未甲基化的dntp进行末端修复时所导致的无法确定突出末端有几个碱基被修复的现象,从而避免了在测序时末端修复的碱基信息也被测序仪计算在内的现象,进而能够避免测序仪读取的碱基数据大于实际的碱基数据的现象,并且克服由于采用完全未甲基化的dntp进行末端修复所导致的数据冗余现象。
[0090]
另外,本技术通过提高双链dna的建库起始量来弥补由于末端切除部分碱基而造成的dna整体数据量降低的现象,使得本技术最终测得的dna甲基化水平非常接近于真实的甲基化水平,所产生的误差在商业化测序时可以忽略不计。
[0091]
总之,本技术采用末端剪切、酶学转化和双链建库法相结合来建立dna甲基化测序文库,不会产生因为采用非甲基化的dntp进行末端修复而造成的数据冗余现象和甲基化水平失真现象,并且,使得所测得的甲基化数据接近于真实的甲基化水平,有效克服了现有的甲基化测序技术的缺陷。
[0092]
以下结合实施例对本技术作进一步的说明。
[0093]
实施例一
[0094]
本实施例提供了一种末端突出的双链dna的获得方法,其包括如下步骤:
[0095]
1、提取目标样品中的双链dna,检测该双链dna的浓度,并且采用电泳检测提取的双链dna条带的分子量是否属于所需的dna的分子量。如果属于,则进行下一步。如果不属于,则重新提取dna。
[0096]
2、可选地,超声打断提取的dna,其具体包括:根据dna的浓度和电泳条带确定dna的上样量,一般打断起始量为150ng,总体积为100μl,置于打断仪中进行打断,将打断后的dna进行2%琼脂糖凝胶电泳检测,dna片段在240bp至290bp之间时,即得含有末端突出的双链dna的dna测序样品。打断仪的程序为打断30s,然后暂停30s,共持续9个循环。
[0097]
当从组织中提取基因组dna时,由于其片段较长,故需要采用上述的打断方法。而从血液中提取到的cfdna的片段一般位于120bp至200bp之间,例如为170bp,故无需上述的打断过程。但如果提取到的dna的片段超过300bp时,也可以进行打断。
[0098]
本技术的技术适用于末端突出的双链dna,例如血液中游离的cfdna、ctdna和一些组织基因组dna等。上述的dna在血液环境中容易被酶消化,从而3’末端的不平整现象要比来自于细胞核的组织dna更常见。
[0099]
实施例二
[0100]
本实施例涉及一种末端切除产物的制备方法,其采用核酸外切酶对实施例一所得的dna产物中的双链dna的突出的5’和/或3’末端进行末端切除,以形成平末端的双链dna。
[0101]
其中,上述的末端切除体系包括:44μl的实施例一所得的dna产物、1μl的核酸外切酶(exonuclease vii)、5μl的10x核酸外切酶缓冲液(10xexonuclease vii buffer)。以上试剂在1个pcr管中混合,最终得到的体积为50μl。然而,上述的体积可以根据dna产物的不
同而改变。
[0102]
末端切除的反应条件为:在37℃的温度条件下反应30min,然后在95℃的温度条件下反应10min,最后在4℃的温度条件下保存。
[0103]
其中,本实施例的核酸外切酶能够从5’和/或3’末端依次降解突出的dna部分。
[0104]
与本实施例不同,如果在建立测序文库时采用未甲基化的dntps来填补末端,由于末端修复的碱基与原有的碱基难以区分,故该种情况下的末端修复会导致原始双链dna的末端碱基缺失数目无法精确测得,这会造成测序所得到的碱基数目失真,并且也会在末端引入新的甲基化水平而造成甲基化信息失真。而本实施例直接切除dna的突出末端,使之成为平头末端的dna,而并未采用dntps来填补末端,因此,本实施例不会引入冗余的序列,也不会引入新的甲基化水平,从而不会造成甲基化信息失真的现象。
[0105]
本实施例虽然切除了突出的末端,造成了数据量的降低,但是该数据量的降低可以通过提高构建文库所用的dna的起始总量来弥补。针对每个末端突出的双链dna,其突出碱基数一般在1-80个碱基之间。但是针对同一来源的双链dna分子而言,每个双链dna分子由于受到体内环境的不同的影响(例如不同的酶、ph值等),其末端突出的碱基数会存在些许差异。例如,在同一来源的单个不同的dna分子中,有的dna分子末端突出的碱基数较少,如为1个碱基、2个碱基、3个碱基、15个碱基等,而有的dna分子的末端突出的碱基数较多,如为80个碱基、70个碱基、65个碱基、60个碱基等。如果提高构建文库所用的dna的起始总量,则同一来源的双链dna分子的数量有了大量增加,即便单个dna分子可能差异较大,但是在大量dna分子存在的前提下,可以用更多的dna分子提高测序深度,这相当于缩小了同一来源的每一个dna分子之间的差异,由此,能够提高甲基化测序的精度。
[0106]
实验证明,针对具有同一来源的170个碱基长度的cfdna,如果突出末端的平均突出长度为15个(即为总碱基长度的8.8%),那么cfdna的起始量要至少调高8.8%,甚至更高。又例如,如果突出末端的平均突出长度为34个(即为总碱基长度的20%),那么cfdna的起始量要至少调高20%,甚至更高。发明人发现,如果dna的起始量的调高程度与dna的末端突出的比例一致,能够基本保证末端切除的量在最终计算甲基化水平时能够得到补偿。dna的建库起始量调得越高,测序覆盖深度越深,越能减少随机抽样带来的波动,最终的甲基化水平会越接近于真实值。但是基于成本和效率的考量,相对于现有的基于酶学转化法的双链建库法通常的dna的起始量而言,本技术的dna的起始量的调高的百分比是双链dna的末端切除率或平均末端切除率的1.3至2倍。本实施例中,双链dna的起始量为30ng,而基于亚硫酸盐转化法的单链建库法或基于酶学转化法的双链建库法的双链dna的起始量一般为20ng,故本实施例的双链dna的起始量高于未剪切末端的建库法所采用的起始量。
[0107]
总之,虽然本实施例的方法通过切除突出末端而不会引入新的甲基化水平,但是又通过提高用于建立测序文库的dna的起始量来弥补由于末端切除而造成的dna样品的整体数据量的损失,所以本实施例的甲基化信息接近于真实值,并且不会造成因为现有技术采用的末端修补法而造成的甲基化信息失真的现象。
[0108]
实施例三
[0109]
本实施例提供了一种dna混合样品的制备方法,其包括如下步骤:
[0110]
1、加入内参dna:
[0111]
在实施例二所得的末端切除产物中加入1μlλdna和1μlpuc19 dna,在0.1x的te和
ph为8.0的条件下混合,然后得到总体积为55μl的dna混合样品。λdna和puc19 dna作为对照dna(control dna),其加入的量根据末端切除产物中双链dna(cfdna或ctdna)的读长(reads)的不同而不同。
[0112]
本实施例中,加入的puc19 dna(cpg methylated puc19)的所有的胞嘧啶(c)是完全甲基化的,puc19的甲基化率用于指示实验中对甲基化胞嘧啶的保护是否有效。如果最终分析结果中,puc19甲基化率大于98%时可以视为甲基化保护成功。因此,加入的puc19 dna可以作为内参dna。
[0113]
本实施例中,加入的λdna(unmethylated lambda dna)是完全未甲基化的,即其上所有的胞嘧啶(c)是完全不甲基化的。λdna的甲基化率可以指示实验中对未甲基化c的转化是否有效。如果最终分析λdna甲基化率低于1.5%时可以视为试验成功。因此,加入的λdna也可以作为内参dna。
[0114]
实施例四
[0115]
本实施例提供了一种接头连接产物的制备方法,其采用em-seq adapter试剂盒在实施例三所得的dna混合样品中的双链dna的两个平头末端添加接头序列(adapter),所得到的待测双链dna即为接头连接产物。
[0116]
本实施例的用于添加接头序列的反应体系包括:50μl实施例三所得的dna混合样品、2.5μl的nebnext em-seq接头序列(adapter)、1μl的连接增强剂(nebnext ligation enhancer)和30μl连接反应混合物(nebnext ultra ii ligation master mix)。将上述试剂加入单个pcr管中,总体积为83.5μl。
[0117]
接头序列由nebnext enzymatic methyl-seq kit试剂盒提供,用于供测序仪读取而实现测序功能。
[0118]
本实施例的反应条件为:将总体积为83.5μl的pcr管置于20℃下反应15min,即得接头连接产物。反应时不使用热盖,反应产物可以视情况进行纯化。
[0119]
实施例五
[0120]
本实施例提供了一种甲基化保护产物的制备方法,其包括如下步骤:
[0121]
1、配制甲基化保护液:
[0122]
将10μl的tet2反应缓冲液(tet2 reaction buffer)、1μl的氧化补剂(oxidation supplement)、1μl的二硫苏糖醇(dithiothreitol,dtt)、1μl的氧化增强剂(oxidation enhancer)和4μl的tet2试剂在pcr薄壁管中混匀,得到17μl的甲基化保护液。
[0123]
其中,使用的试剂盒为neb公司货号为e7125的酶法甲基化转化模块试剂盒,即enzymatic methyl-seq conversion module。
[0124]
tet2酶和氧化增强剂作为甲基化保护试剂用于保护甲基化胞嘧啶上的甲基,其能够通过化学反应改变甲基基团,使其对脱氨试剂惰性,而未甲基化的胞嘧啶上不存在甲基,故不会被氧化而仍然以胞嘧啶形式存在于dna中。dna上的甲基化胞嘧啶包括5-甲基胞嘧啶(5-methylcytosine,5mc)或5-羟甲基胞嘧啶(5-hydroxymethylcytosine,5hmc)。5-羟甲基胞嘧啶是5-甲基胞嘧啶的羟基化形式。tet2酶(ten eleven translocation)属于双加氧酶家族,其结构包括一个保守的c端的催化区和一个n端的调节区。c端的催化区为fe
2+
和/或α-酮戊二酸(α-kg)依赖型,是氧化5mc或5hmc的催化结构域。tet2酶和氧化增强剂的共同作用能够将5-甲基胞嘧啶(5mc)氧化为5-羟甲基胞嘧啶(5hmc),并将5-羟甲基胞嘧啶(5hmc)氧
化为5-甲酰基胞嘧啶(5-formylcytosine,5fc)或5-羧基胞嘧啶(5-carboxylcytosine,5cac),这相当于在胞嘧啶上加上了甲酰基或羧基等保护基团,从而对脱氨试剂表现出惰性,在后继的反应过程中使得胞嘧啶不会由于发生脱氨而生成尿嘧啶。与5mc和5hmc相比,5-甲酰基胞嘧啶和5-羧基胞嘧啶在哺乳动物基因组中含量极低。氧化增强剂用于增加tet2酶的活性。
[0125]
2、进行氧化反应,具体包括:
[0126]
(2-1)、将总体积为28μl的实施例四所得的接头连接产物与17μl的甲基化保护液相混合,加入5μl二价铁稀释液(diluted fe(ii)solution),总体积为50μl/管,立即震荡混匀和短暂离心,置于pcr仪中,在37℃反应1h。
[0127]
(2-2)、反应结束后,将反应液置于冰盒上,加入1μl的终止试剂(stop reagent),震荡混匀后短时离心。
[0128]
(2-3)、将反应液置于pcr仪中,使用热盖,热盖的温度≥45℃,在37℃的温度下反应0.5h,此时,氧化反应已经进行完毕,得到了甲基受保护的双链dna,即甲基化保护产物。
[0129]
其中,上述的二价铁稀释液的配制方法包括:取1μl 500mm二价铁溶液(fe ii solution),加入到1249μl nf水中。因为二价铁溶液容易被氧化,故需要在使用前配制,现配现用。二价铁离子是tet2酶的催化区所依赖的金属离子,为了最大化tet2酶的催化活性,故需要在氧化反应体系中加入二价铁离子。
[0130]
可选地,在氧化反应后对上述的甲基化保护产物进行纯化,得到纯化后的dna。
[0131]
实施例六
[0132]
本实施例提供了一种对实施例五所得的甲基化保护产物或纯化后的dna(当采取纯化步骤对甲基化保护产物进行纯化时)进行变性的方法,变性的目的是使上述的双链dna变性为单链dna,即变性产物,具体包括如下步骤:
[0133]
取16μl实施例五所得的甲基化保护产物或纯化后的dna,置于pcr反应管中,加入4μlnaoh(0.1mol/l),放入预热后的pcr仪中,盖上热盖(温度≥60℃),50℃孵育10min,孵育结束后,立即取下反应管,置于冰盒上保存。
[0134]
其中,naoh作为变性剂用于使得待测双链dna变性为单链dna。
[0135]
实施例七
[0136]
本实施例提供了一种单链dna的转化方法,其包括如下步骤:
[0137]
1、配制脱氨反应液
[0138]
取1支pcr薄壁管,加入10μlapobec反应缓冲液(apobec reaction buffer)、1μl牛血清白蛋白(bovine serum albumin,bsa)、1μlapobec,然后加入68μl无核酸酶水(nuclease-free water,nf water),充分震荡混匀后,配制成80μl脱氨反应液。
[0139]
2、进行脱氨反应
[0140]
用移液器吸取80μl脱氨反应液置于实施例六所得的20μl变性产物(即已变性完成的单链dna)中,充分震荡混匀,并置于预热好的pcr仪上,并盖上热盖(温度≥45℃),于37℃孵育3h。
[0141]
在该步骤中,apobec蛋白的c端具有胞嘧啶脱氨酶区,能够使胞嘧啶脱去氨基变成尿嘧啶,因此,apobec作为胞嘧啶脱氨酶将实施例六所得的单链dna中未甲基化的胞嘧啶(c)转化为尿嘧啶(u),而受保护胞嘧啶不能被apobec蛋白脱氨,故不会转化为尿嘧啶。脱氨
反应后,得到了转化后单链dna。
[0142]
3、在脱氨后进行纯化,得到能够用于pcr扩增的转化后的单链dna。
[0143]
实施例八
[0144]
本实施例提供了一种利用转化后单链dna构建dna甲基化测序文库的方法,其包括如下的pcr扩增步骤:
[0145]
取pcr管,加入20μl上述的转化后单链dna,加入5μl em-seq index primer plate和25μlnebnext q5u master mix,进行pcr反应,pcr的程序为先在98℃时运行30s;然后在98℃时持续10s,62℃时持续30s,64℃时持续60s,一共持续9个循环;65℃时持续5min,最后在4℃时保存。
[0146]
经过上述的pcr扩增之后,u碱基的配对碱基是a,a碱基配对碱基是t。因为pcr过程中使用的寡核苷酸是a、t、c、g,不含u,在经过n个循环(n≥9)过后,含u的原始链变成了仅有1/(29),可以忽略不计。由此,双链dna中的尿嘧啶(u)被转化为胸腺嘧啶(t)。
[0147]
在pcr反应完毕后,得到能够被测序仪读取的dna甲基化测序文库。
[0148]
实施例九
[0149]
本实施例提供了一种dna甲基化水平的检测方法,其包括如下步骤:
[0150]
1、对5’和/或3’末端突出的双链dna按照上述各个实施例提供的制备方法构建dna甲基化测序文库;
[0151]
2、采用测序仪对dna甲基化测序文库进行测序,得到测序结果;
[0152]
3、对测序结果进行分析,得到5’末端突出的双链dna的甲基化水平。
[0153]
上述步骤的序号并不视为对上述步骤的顺序的唯一限定。
[0154]
实验例
[0155]
本实验例对本技术的测序文库与现有的商用试剂盒采用的酶学法甲基化实验构建的测序文库的测序结果进行对比,其结果如图5所示。图5是本技术的方案相对现有技术的结果对比图。现有技术包括如下步骤:先在双链状态下使用未甲基化的dntp进行末端修复,再加多a序列,加接头,进行酶学转化,并完成pcr扩增,从而得到甲基化测序文库。
[0156]
在图5中,read2通过illumina公司的novaseq600型号测序仪读取平均甲基化率变化。纵坐标表示read2在各读取位置上的平均甲基化率,横坐标从左至右的方向表示读长2(read 2)从p5端至p7端的方向,横坐标的值表示测序仪读取的碱基位置。图5中的上方曲线表示本技术建立的文库测得的甲基化率,下方曲线表示现有酶学法建立的文库测得的甲基化率。本实验例的样本与现有酶学法所使用的样本相同。
[0157]
进入测序仪进行测序的每个dna小片段的长度约在200至300个碱基之间,甚至可能更短。测序仪在读每个dna小片段的时候,并不能从头读到尾,只能从头读150个碱基的长度,并从尾读150个碱基的长度。即测序仪分别从dna片段的两头读取,各读150个碱基长度,故把这些测序仪读取产生的短读段称为reads,单个短读段称为read。每条文库dna单链在二代测序仪上读取的时候,是先从p5端到p7端读150碱基,为read1;再从p7端到p5端读150个碱基,为read2。p5和p7是illumina测序仪的芯片上的接头名称,p5对应5'端,p7对应3'端。测序仪的芯片上的接头与测序文库的dna分子上的接头序列有互补性。
[0158]
从图5可知,针对相同样本,现有酶学法测得的平均甲基化率在1至29个碱基范围内与本技术测得的平均甲基化率基本相同,在29个碱基到131个碱基范围内存在显著不同。
理论上,因为所有待测序分子在基因组上的位置分布是随机的,因此其平均甲基化率不应该随读取位置而变化。但是从图5可以看到,现有酶学法在越接近p7端(即进行末端修复的3’端方向)时,平均甲基化率越低,说明末端修复引入的未甲基化dntp对样本原始甲基化信号造成了污染,且在越靠近原始单链的3’端时,这种污染越明显。而本技术的方法则避免了这个问题,在各个读取位置的平均甲基化率上保持了良好的一致性。这能够说明:与现有的基于酶学转化法的双链建库法相比,本技术不会在末端引入新的甲基化水平,不会产生甲基化水平失真或降低现象。
[0159]
以上对本技术进行了详细介绍,本文中应用了具体个例对本技术的原理及实施方式进行了阐述,以上实施例的说明只是用于帮助理解本技术的方法及其核心思想;同时,对于本领域的技术人员,依据本技术的思想,在具体实施方式及应用范围上均会有改变之处,综上,本说明书内容不应理解为对本技术的限制。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1