一种富马酸卢帕他定中间体及富马酸卢帕他定的制备方法与流程
1.本发明属于化学合成技术领域,具体涉及一种富马酸卢帕他定中间体及富马酸卢帕他定的制备方法。
背景技术:
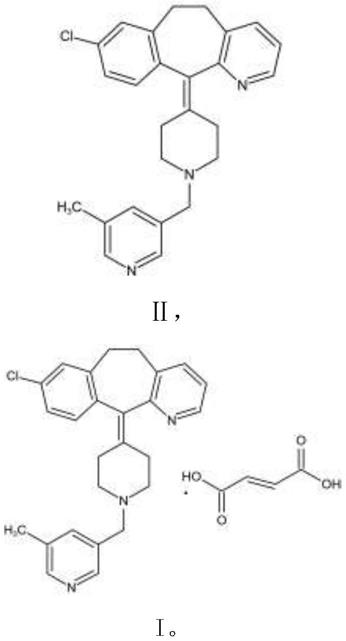
2.富马酸卢帕他定(rupatadine fumarate),化学名为8-氯-11-[1-[(5-甲基-3-吡啶基)甲基]-4-哌啶亚基]-6,11-二氢-5h-苯并[5,6]环庚并[1,2-b]吡啶富马酸盐,西班牙 uriach公司研制的具有抗组胺和血小板活化因子(paf)双重作用的抗过敏药物,适应症为季节性和常年性过敏性鼻炎。富马酸卢帕他定的化学结构式如下所示:
[0003]
在synthetic communications,38(1),122-127;2008中提到一种富马酸卢帕他定的制备方法,具体为:
[0004]
该方法的缺点在于:在反应中使用了四氢铝锂,反应温度在-70~-75℃以下,危险性大,且反应条件苛刻,不适合工业化生产。酯化与氯代均需使用二氯亚砜,对环境污染严重。
[0005]
在《卢帕他定的合成》(辛水波,中国新药杂志,2005年)中公开了一种富马酸卢帕他定的合成路线,具体为:
[0006]
该方法的缺点在于:溴代反应收率低,且溴代过程中存在多取代现象,难以分离,需要柱层析纯化,收率低,后处理复杂,不适合工业化生产。
[0007]
在《富马酸卢帕他定的合成》(陈建华,中国医药工业杂志,2007)中也公开了一种富马酸卢帕他定的合成路线,具体为:
[0008]
该方法的缺点在于:酰化反应用到dcc,副产物dcu很难除去,还原用到pocl3,会生成大量胶体状物,搅拌不动,副产物多,后处理复杂,收率低,不适合商业化生产。
[0009]
综上所述,现有制备富马酸卢帕他定或其中间物的合成均线存在副产物多,收率低,后处理复杂等技术问题。
技术实现要素:
[0010]
有鉴于此,本发明目的在于提供一种富马酸卢帕他定中间体的制备方法,所述富马酸卢帕他定中间体的结构式如式
ⅴ
所示,现有技术缺陷也主要是在该中间体的合成上存在技术缺陷。该方法在合成富马酸卢帕他定中间体上反应条件温和,后处理不复杂,且收率高。
[0011]
其中,式
ⅴ
中,r1选自cl或br。
[0012]
所述富马酸卢帕他定中间体的制备方法包括:将式ⅵ所示化合物与卤代试剂反应
得到所述式
ⅴ
所示的中间体;
[0013]
具体地,式ⅵ化合物在卤代试剂与溶剂存在下反应得到式(
ⅴ
)化合物的合成路线如下所示:
[0014]
其中,3,5-二甲基吡啶-n-氧化物中n—o结构片段,既有σ吸电子效应,同时还具有π体系电子反馈作用,在特殊电子效应作用下3,5-二甲基吡啶-n-氧化物活性远高于 3,5-二甲基吡啶,与卤代试剂反应更易进行,且避免了卤代产物与吡啶氮原子发生副反应生成季铵盐。
[0015]
进一步,所述卤代试剂选自n-溴代丁二酰亚胺(nbs)、n-氯代丁二酰亚胺(ncs) 中的一种,优选为n-溴代丁二酰亚胺(nbs)。
[0016]
进一步,所述式ⅵ化合物与所述卤代试剂的摩尔比为1:1-1.5。优选为1:1.2。
[0017]
进一步,所述反应加入引发剂进行反应,所述引发剂为偶氮二异丁腈(aibn)。
[0018]
进一步,所述式ⅵ化合物与所述引发剂摩尔比为1:0.05-0.1,优选为1:0.1。
[0019]
进一步,所述反应的反应溶剂为三氯甲烷、二氯甲烷、乙腈中的一种或多种混合;优选为乙腈。
[0020]
进一步,所述反应在65℃-反应溶剂回流温度范围内进行,优选为在溶剂回流温度下反应。
[0021]
进一步,将所述式
ⅴ
所示的中间体与式ⅳ所示化合物加入缚酸剂进行反应得式ⅲ所示化合物;所述反应在0℃-反应溶剂回流温度范围内进行;
[0022]
具体地,式
ⅴ
化合物在式ⅳ化合物与溶剂存在下反应得到式ⅲ化合物的合成路线如下所示:
[0023]
进一步,反应得式ⅲ所示化合物的反应溶剂为二氯甲烷、thf、甲苯、乙酸乙酯中的一种或多种混合。
[0024]
进一步,所述式ⅳ化合物与所述式
ⅴ
化合物的摩尔比为1:1.2-1.5。
[0025]
进一步,所述式ⅳ化合物与所述缚酸剂的摩尔比为1:2-2.5,优选为1:2。
[0026]
进一步,所述缚酸剂为三乙胺、碳酸钠中的一种,优选三乙胺。
[0027]
进一步,所述反应的反应溶剂为二氯甲烷、thf、甲苯、乙酸乙酯中的一种或多种混合,优选乙酸乙酯。
[0028]
进一步,所述反应在0℃-溶剂回流温度范围内进行。反应温度优选为10℃-20℃。
[0029]
进一步,所述反应得式ⅲ所示化合物经监控式ⅳ化合物消失后,加ph值4-6的缓冲液进行洗涤、浓缩;然后加乙酸乙酯与正己烷的混合溶液精制得到精制式ⅲ所示化合物;所述缓冲液为氢氧化钠、水和乙酸的混合溶液。
[0030]
本发明在制备富马酸卢帕他定的基础上,进一步提供一种富马酸卢帕他定的制备
方法,其结构式如式i所示,所述制备方法包括:(1)使用前任一所述的制备方法制备得到的式ⅲ所示化合物与还原剂反应得式ⅱ所示化合物;(2)将所述式ⅱ所示化合物于富马酸反应得到所述富马酸卢帕他定;
[0031]
具体地,所述步骤(1)合成路线如下所示:
[0032]
具体地,所述步骤(2)合成路线如下所示:
[0033]
进一步,所述步骤(1)中,所述还原剂为铁粉与酸、tct(三聚氯氰)中的一种,优选tct(三聚氯氰)。
[0034]
进一步,所述步骤(1)中,所述式ⅲ化合物与所述还原剂摩尔比为1:1-1.5。优选为优选1:1.1。
[0035]
进一步,所述步骤(1)中,所述反应在10℃-40℃温度范围内进行,反应温度优选为20℃-30℃。
[0036]
进一步,所述步骤(1)中,所述反应的反应溶剂为thf、乙腈、甲苯、二氯甲烷、丙酮中的一种或多种混合,优选乙腈。
[0037]
进一步,所述步骤(2)中,所述反应的反应溶剂为乙酸乙酯、乙醇、二氯甲烷、水、丙酮中的一种,优选丙酮或乙酸乙酯,更优选丙酮与乙酸乙酯混合溶剂。
[0038]
进一步,所述步骤(2)中,所述反应在0℃-溶剂回流温度范围内进行,反应温度优选为溶剂回流温度。
[0039]
进一步,所述步骤(2)反应完后,将反应液混合物冷却降温至0-10℃,过滤、洗涤、干燥得到式ⅰ化合物。
[0040]
具体地,式ⅵ所示化合物到富马酸卢帕他定的合成路线从下所示:本发明有益效果在于
[0041]
本发明提供的富马酸卢帕他定中间体(化合物v)的制备方法用3,5-二甲基吡啶-n
‑ꢀ
氧化物进行溴代或氯代反应,与用3,5-二甲基吡啶进行溴代或氯代反应比较,本发明卤代产物纯度和收率更高。
[0042]
本发明提供的富马酸卢帕他定中间体(化合物ⅲ)的制备方法,式
ⅴ
化合物与式ⅳ化合物反应后,不需要柱层析,采用溶剂精制得到式(ⅲ)化合物,后处理操作简单,更有利于工业化生产。
[0043]
本发明提供的富马酸卢帕他定中间体的制备方法及制备富马酸卢帕他定的方法工艺简单,反应温度适中,无超低温或高温反应,便于工业化生产,所用溶剂或试剂便宜易得,加料方便,运输储存简单,而且,精制溶剂体系除杂能力强,收率高,适合商业化生产。
附图说明
[0044]
图1为实例1制备的化合物
ⅴ
的高效色相色谱图。
[0045]
图2为实例2制备的化合物
ⅴ
的高效色相色谱图。
[0046]
图3为实例6制备的化合物ⅲ的高效色相色谱图。
[0047]
图4为实例9制备的化合物ⅱ的高效色相色谱图。
[0048]
图5为实例12制备的化合物ⅰ的高效色相色谱图。
[0049]
图6为实例1制备的化合物
ⅴ
的质谱图。
[0050]
图7为实例6制备的化合物ⅲ的质谱图。
[0051]
图8为实例9制备的化合物ⅱ的质谱图。
[0052]
图9为实例12制备的化合物ⅰ的红外光谱图。
[0053]
图10为实例12制备的化合物ⅰ的质谱图。
[0054]
图11为实例12制备的化合物ⅰ的核磁共振氢谱图。
[0055]
图12为实例12制备的化合物ⅰ的核磁共振碳谱图。
具体实施方式
[0056]
所举实施例是为了更好地对本发明进行说明,但并不是本发明的内容仅局限于所举实施例。所以熟悉本领域的技术人员根据上述发明内容对实施方案进行非本质的改进和调整,仍属于本发明的保护范围。
[0057]
本发明实施例中,高效液相色谱测试的条件为:
[0058]
本发明实施例中,质谱测试选择:ab公司的api3000,esi源。
[0059]
本发明实施例中,红外光谱测试的测试仪器选择shimadzu ftir-8400红外分光光度计,测试方法选择溴化钾压片法。
[0060]
实例1化合物
ⅴ
制备
[0061]
氮气保护下,将式(ⅵ)化合物(2.5g,0.02mol,1.0eq)加入100ml反应瓶中,加入乙腈50ml,开启搅拌,升温至回流,加入ncs(3.2g,0.024mol,1.2eq)、aibn(0.33g, 0.002mol,0.1eq),反应6h,tlc(展开剂,甲醇:二氯甲烷=1:2,加2滴氨水)监控式(ⅵ) 化合物消失,减压浓缩掉乙腈,加甲苯20ml搅拌30min,过滤,收集滤液,室温搅拌下滴加正己烷60ml,滴加完毕,降温至0-10℃搅拌60min,过滤,干燥得到式(
ⅴ
)化合物(2.8g),收率88.9%,hplc纯度86.05%,见图1。ms-esi(m/z):157.1[m+h]
+
,见图6。
[0062]
实例2化合物
ⅴ
制备
[0063]
氮气保护下,将式(ⅵ)化合物(2.5g,0.02mol,1.0eq)加入100ml反应瓶中,加入乙腈50ml,开启搅拌,升温至回流,加入ncs(4.0g,0.03mol,1.5eq)、aibn(0.33g, 0.002mol,0.1eq),反应6h,tlc监控式(ⅵ)化合物消失,减压浓缩掉乙腈,加甲苯20ml 搅拌30min,过滤,收集滤液,室温搅拌下滴加正己烷60ml,滴加完毕,降温至0-10℃搅拌 60min,过滤,干燥得到式(
ⅴ
)化合物(2.6g),收率82.5%,hplc纯度67.05%,见图2。
[0064]
实例3化合物
ⅴ
制备
[0065]
氮气保护下,将式(ⅵ)化合物(2.5g,0.02mol,1.0eq)加入100ml反应瓶中,加入乙腈50ml,开启搅拌,升温至回流,加入ncs(2.67g,0.02mol,1.0eq)、aibn(0.33g, 0.002mol,0.1eq),反应6h,tlc监控式(ⅵ)化合物未反应完。
[0066]
实例4化合物
ⅴ
制备
[0067]
氮气保护下,将式(ⅵ)化合物(2.5g,0.02mol,1.0eq)加入100ml反应瓶中,加入二氯甲烷50ml,开启搅拌,升温至回流,加入ncs(3.2g,0.024mol,1.2eq)、aibn(0.33g, 0.002mol,0.1eq),反应6h,tlc监控式(ⅵ)化合物未反应完。
[0068]
实例5化合物
ⅴ
制备
[0069]
氮气保护下,将式(ⅵ)化合物(2.5g,0.02mol,1.0eq)加入100ml反应瓶中,加入三氯甲烷50ml,开启搅拌,升温至回流,加入ncs(3.2g,0.024mol,1.2eq)、aibn(0.33g,0.002mol,0.1eq),反应6h,tlc监控式(ⅵ)化合物未反应完。
[0070]
实例6化合物ⅲ制备
[0071]
氮气保护下,将式(
ⅴ
)化合物(2.4g,0.015mol,1.25eq)加入100ml反应瓶中,加入乙酸乙酯20ml,开启搅拌,加入式(ⅳ)化合物(3.7g,0.012mol,1.0eq),控制反应液温度在10-20℃,加入三乙胺(2.4g,0.024mol,2.0eq),反应5h,tlc(展开剂,甲醇: 乙酸乙酯=1:2,加2滴氨水)监控式(ⅳ)化合物消失,加ph值4-6的缓冲液(用氢氧化钠、水、乙酸,混合配制)洗涤两次,室温下滴加正己烷20ml,滴加完毕,降温至0-10℃搅拌120min,过滤,干燥得到式(ⅲ)化合物(4.8g),收率92.3%,hplc纯度98.83%,见图3。ms-esi(m/z):432.2[m+h]
+
,见图7。
[0072]
实例7化合物ⅲ制备
[0073]
氮气保护下,将式(
ⅴ
)化合物(2.8g,0.018mol,1.5eq)加入100ml反应瓶中,加入乙酸乙酯20ml,开启搅拌,加入式(ⅳ)化合物(3.7g,0.012mol,1.0eq),控制反应液温度在10-20℃,加入三乙胺(2.4g,0.024mol,2.0eq),反应5h,tlc(展开剂,甲醇: 乙酸乙酯=1:2,加2滴氨水)监控式(ⅳ)化合物消失,加ph值4-6的缓冲液(用氢氧化钠、水、乙酸,混合配制)洗涤两次,室温下滴加正己烷20ml,滴加完毕,降温至0-10℃搅拌120min,过滤,干燥得到式(ⅲ)化合物(4.81g),收率92.5%。
[0074]
实例8化合物ⅲ制备
[0075]
氮气保护下,将式(
ⅴ
)化合物(1.9g,0.012mol,1.0eq)加入100ml反应瓶中,加入乙酸乙酯20ml,开启搅拌,加入式(ⅳ)化合物(3.7g,0.012mol,1.0eq),控制反应液温度在10-20℃,加入三乙胺(2.4g,0.024mol,2.0eq),反应5h,tlc(展开剂,甲醇: 乙酸乙酯=1:2,加2滴氨水)监控式(ⅳ)化合物未反应完。
[0076]
实例9化合物ⅱ制备
[0077]
氮气保护下,将式(ⅲ)化合物(4.0g,0.0093mol,1.0eq)加入100ml反应瓶中,加入乙腈60ml,开启搅拌,分批加入tct(1.9g,0.01mol,1.1eq),控制室温反应,tlc(展开剂,甲醇:乙酸乙酯=1:2,加2滴氨水)监控式(ⅲ)化合物消失,向反应液滴加50ml饱和碳酸钠溶液,搅拌30min。加入二氯甲烷提取,有机相用水洗,减压浓缩除去溶剂,加入乙酸乙酯10ml回流溶解,加入正己烷30ml搅拌30min,降温至0-10℃搅拌60-90min,过滤、干燥得到式(ⅱ)化合物(3.6g),收率92.3%,hplc纯度99.61%,见图4。ms-esi(m/z): 417.9[m+h]
+
,见图8。
[0078]
实例10化合物ⅱ制备
[0079]
氮气保护下,将式(ⅲ)化合物(4.0g,0.0093mol,1.0eq)加入100ml反应瓶中,加入甲苯60ml,开启搅拌,分批加入tct(1.9g,0.01mol,1.1eq),控制室温反应,tlc(展开剂,甲醇:乙酸乙酯=1:2,加2滴氨水)监控式(ⅲ)化合物消失,向反应液滴加50ml饱和碳酸钠溶
液,搅拌30min。加入二氯甲烷提取,有机相用水洗,减压浓缩除去溶剂,加入乙酸乙酯10ml回流溶解,加入正己烷30ml搅拌30min,降温至0-10℃搅拌60-90min,过滤、干燥得到式(ⅱ)化合物(3.1g),收率79.5%。
[0080]
实例11化合物ⅱ制备
[0081]
氮气保护下,将式(ⅲ)化合物(4.0g,0.0093mol,1.0eq)加入100ml反应瓶中,加入thf 60ml,开启搅拌,分批加入tct(1.9g,0.01mol,1.1eq),控制室温反应,tlc(展开剂,甲醇:乙酸乙酯=1:2,加2滴氨水)监控式(ⅲ)化合物消失,向反应液滴加50ml饱和碳酸钠溶液,搅拌30min。加入二氯甲烷提取,有机相用水洗,减压浓缩除去溶剂,加入乙酸乙酯10ml回流溶解,加入正己烷30ml搅拌30min,降温至0-10℃搅拌60-90min,过滤、干燥得到式(ⅱ)化合物(2.9g),收率74.4%。
[0082]
实例12化合物i制备
[0083]
氮气保护下,将式(ⅱ)化合物(35g,0.084mol,1.0eq)加入2000ml反应瓶中,加入丙酮350ml,开启搅拌,升温至溶剂回流,固体溶解完全后加入富马酸(9.8g,0.084mol, 1.0eq),搅拌反应30min后,加入乙酸乙酯350ml,降温至室温搅拌30min,再降温至0-10℃搅拌60-90min,过滤、干燥得到式(ⅰ)化合物(43.98g),收率98.2%,hplc纯度99.76%,见图5。ir(红外光谱),见图9。ms-esi(m/z):530.3[m-1h]-,见图10。1h-nmr(核磁共振氢谱),见图11。
13
c-nmr(核磁共振碳谱),见图12。
[0084]
实例13化合物i制备
[0085]
氮气保护下,将式(ⅱ)化合物(5g,0.012mol,1.0eq)加入250ml反应瓶中,加入无水乙醇50ml,开启搅拌,升温至溶剂回流,固体溶解完全后加入富马酸(1.4g,0.012mol,1.0eq),搅拌反应30min后,降温至室温搅拌30min,再降温至0-10℃搅拌 60-90min,过滤、干燥得到式(ⅰ)化合物(5.94g),收率92.96%。
[0086]
实例14化合物i制备
[0087]
氮气保护下,将式(ⅱ)化合物(5g,0.012mol,1.0eq)加入250ml反应瓶中,加入丙酮50ml,开启搅拌,升温至溶剂回流,固体溶解完全后加入富马酸(1.4g,0.012mol, 1.0eq),搅拌反应30min后,降温至室温搅拌30min,再降温至0-10℃搅拌60-90min,过滤、干燥得到式(ⅰ)化合物(6.1g),收率95.6%。
[0088]
实施例15实施例1制备的化合物
ⅴ
测试
[0089]
将实施例1制备的化合物
ⅴ
进行液相色谱测试,hplc(液相色谱)纯度86.05%,液相色谱积分结果如表1,色谱图如图1所示。表1hplc测定化合物
ⅴ
纯度积分结果表
[0090]
将实施例1制备的化合物
ⅴ
进行质谱测试(m/z(质谱):仪器型号:ab公司的api3000, esi源),测试数据:157.1[m+h]
+
(如图6所示)。
[0091]
实施例16实施例2制备的化合物
ⅴ
测试
[0092]
将实施例2制备的化合物
ⅴ
进行液相色谱测试。hplc(液相色谱)纯度67.05%,液相色谱积分结果如表2,色谱图如图2所示。表2hplc测定化合物(3-氯甲基-5-甲基吡啶)纯度积分结果表
[0093]
实施例17实施例6制备的化合物ⅲ测试
[0094]
将实施例6制备的化合物ⅲ进行液相色谱,hplc(液相色谱)纯度98.83%,液相色谱积分结果如表3,色谱图如图3所示。表3hplc测定化合物ⅲ纯度积分结果表
[0095]
将实施例6制备的化合物ⅲ进行质谱测试(m/z(质谱):仪器型号:ab公司的api3000, esi源),测试数据:432.2[m+h]
+
(如图7所示)。
[0096]
实施例18实施例9制备的化合物ⅱ测试
[0097]
将实施例9制备的化合物ⅱ进行液相色谱,hplc(液相色谱)纯度99.61%,液相色谱积分结果如表4,色谱图如图4所示。表4hplc测定化合物ⅱ纯度积分结果表
[0098]
将实施例9制备的化合物ⅱ进行质谱测试,m/z(质谱):仪器型号:ab公司的api3000, esi源,测试数据:417.9[m+h]
+
(如图8所示)。
[0099]
实施例19实施例12制备的化合物ⅰ测试
[0100]
将实施例12制备的化合物ⅰ进行液相色谱,hplc(液相色谱)纯度99.76%,液相色谱积分结果如表5,色谱图如图5所示。
[0101]
表5hplc测定化合物ⅰ纯度积分结果表
[0102]
将实施例12制备的化合物ⅰ进行ir(红外光谱)测试,测试结果:δ=3406cm-1、 3032cm-1、2977cm-1、2923cm-1、2898cm-1、1700cm-1、1662cm-1、1653cm-1、1594cm-1、 1559cm-1、1480cm-1、1438cm-1、1421cm-1、1372cm-1、1164cm-1、1105cm-1、1096cm-1、989cm-1、972cm-1、949cm-1、875cm-1、831cm-1、819cm-1、712cm-1、643cm-1(如图9所示)。
[0103]
将实施例12制备的化合物ⅰ进行质谱测试,测试结果:530.3[m-h]-(如图10所示)。
[0104]
将实施例12制备的化合物ⅰ进行核磁共振氢谱测试,1h-nmr(核磁共振氢谱) (600mhz,dmso)测试结果:δ=8.34-8.32(dd,1h);8.30-8.30(d,1h);8.29-8.28(d,1h);7.58-7.56(dd,2h);7.53(s,1h);7.29-7.29(d,1h);7.21-7.17(m,2h);7.07-7.05(d,1h); 6.62(s,2h);3.51(s,2h);3.35-3.24(m,2h);2.84-2.81(m,2h);2.65-2.62(m,2h); 2.36-2.34(m,2h);2.28(s,3h);2.26-2.23(m,2h);2.20-2.16(m,2h)(如图11所示)。
[0105]
将实施例12制备的化合物ⅰ进行核磁共振氢谱测试,
13
c-nmr(核磁共振碳谱) (600mhz,cd3od)测试结果:δ=168.71;156.23;149.51;147.55;145.72;139.91;139.55; 138.53;136.70;134.57;134.46;134.37;134.27;133.13;130.05;128.99;128.58; 125.83;123.00;57.68;52.93;31.07;30.62;28.35;28.22;16.86(如图12所示)。
[0106]
最后说明的是,以上实施例仅用以说明本发明的技术方案而非限制,尽管参照较佳实施例对本发明进行了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的宗旨和范围,其均应涵盖在本
发明的权利要求范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1