一种重组胶原蛋白双相凝胶及其制备方法和应用与流程
1.本发明涉及生物医用材料技术领域,具体涉及一种重组胶原蛋白双相凝胶及其制备方法和应用。
背景技术:
2.随着年龄或光老化,皮肤会变得松弛或形成皱褶,形成鼻唇沟、鱼尾纹等皱纹或凹陷。软组织填充可以用于修正皱纹或填充凹陷。目前应用于临床的软组织填充材料主要有透明质酸类、羟基磷灰石类、聚乳酸类、聚甲基丙烯酸甲酯类、胶原类等,其中胶原类是最为理想的填充材料。胶原是人体组织的重要组成部分,组织相容性较好,植入后无明显的异物感;其特有的细胞粘附性及细胞增殖诱导作用,使得长期植入后可诱导填充部位细胞增殖而实现组织修复。
3.目前市场上获批的胶原类产品主要有中国台湾双美以猪皮胶原蛋白为原料的交联或非交联胶原蛋白注射剂;美国巴菲特面部整容外科中心的牛皮提取胶原蛋白产品“zyplast”、“zyderm”,以及荷兰hafrod的“爱贝芙”—聚甲基丙烯酸甲酯(pmma)和胶原的复合注射剂。但上述产品仍存在以下诸多技术问题:1、胶原原材料主要来源于动物组织,如牛跟腱,猪皮等,存在病毒、免疫原性风险;2、非永久性填充产品普遍在体内降解过快、需要多次补针,增加消费者负担;3、体内永久植入性产品(如爱贝芙),一方面其中的胶原组分逐渐降解减少、需要反复注射使聚合物微粒数量始终保持在一半以上,才能达到较为长久的美容效果,另一方面不可降解pmma微球存在异物肉芽肿风险,而且它还会在皮肤深层形成晶体无法降解,异物刺激和生物相容性较差。
4.而在对胶原蛋白凝胶双相体系技术上,目前也有相关公开技术;如专利公开号cn103834053a公开了一种可注射的交联透明质酸凝胶及其制备方法,其采用乳化交联获得粒径大于250μm的微球,其将初步交联所得凝胶干燥后复溶、再加入交联剂混合,再经过乳化分散,该方法在乳化之前再次加入交联剂混合,会导致已交联的透明质酸再次发生交联,产生板块状、增加后续乳化压力,交联度较难控制,颗粒粒径分布范围广,且大于250μm的微球具有强烈的异物感;专利公开号cn111840638a公开了一种注射用交联透明质酸填充剂的制备方法,但上述方法中水相的制备需要在2~8℃的低温条件下进行,工艺条件苛刻,且水相混合溶液经乳化后将大量交联剂包裹于微球中,很难清洗彻底,存在安全性风险;另一方面填充剂的制备采用未交联透明质酸作为载体与透明质酸微球共混,载体相易降解,整体体积快速减少、需补充以达到理想的体积支撑效果。
5.由此可见,目前公开的胶原蛋白凝胶填充剂仍存在一定缺点,尚有可以改进的空间。
技术实现要素:
6.本发明目的在于为克服现有的技术缺陷,提供一种重组胶原蛋白双相凝胶及其制备方法和应用。
7.为了解决上述技术问题,本发明提供了以下技术方案:
8.第一方面,提供了一种重组胶原蛋白双相凝胶的制备方法,包括以下步骤:
9.s1:室温下,将重组胶原蛋白a溶解于缓冲溶液中,得到重组胶原蛋白a 溶液;
10.s2:以重组胶原蛋白a溶液为水相,加入到含有表面活性剂的油相中,边搅拌边滴加第一交联剂,在转速600~3000rpm下交联20~60min,得到油包水乳状液;
11.s3:将油包水乳状液离心、清洗、真空干燥,得到重组胶原蛋白a微球;
12.s4:室温下,将重组胶原蛋白b溶解于缓冲溶液中,搅拌得到重组胶原蛋白b凝胶;
13.s5:将重组胶原蛋白a微球加入到重组胶原蛋白b凝胶中,边搅拌边滴加第二交联剂,在转速400~3000rpm下交联1~3h,使重组胶原蛋白a微球均匀分散于重组胶原蛋白b凝胶中,透析去除残留交联剂、脱水,得到重组胶原蛋白双相凝胶;
14.所述重组胶原蛋白a、所述重组胶原蛋白b均选自重组i型人源化胶原蛋白、重组iii型人源化胶原蛋白、重组i型类人胶原蛋白、重组iii型类人胶原蛋白中的一种。
15.进一步地,所述重组胶原蛋白a溶液的浓度为10~200mg/ml;优选地,所述重组胶原蛋白a溶液的浓度为28~100mg/ml。
16.进一步地,所述重组胶原蛋白b凝胶的浓度为10~300mg/ml;优选地,所述重组胶原蛋白b凝胶的浓度为35~200mg/ml。
17.优选地,所述重组胶原蛋白a溶液、重组胶原蛋白b凝胶的ph均为5.0~7.0。
18.进一步地,所述重组胶原蛋白双相凝胶中重组胶原蛋白a微球的质量分数为25-60%。
19.进一步地,所述重组胶原蛋白a微球的粒径为20~200μm,优选地,粒径为40~70μm。
20.进一步地,所述重组胶原蛋白b的分子量大于重组胶原蛋白a的分子量。
21.优选地,所述重组胶原蛋白a的分子量为10~130kda;所述重组胶原蛋白 b的分子量为20~200kda。
22.进一步地,步骤s2中,所述油相为液体石蜡、硅油、植物油、医用白油中的一种;所述表面活性剂为span-80、span-80和tween-60的复配液、abilem90中的至少一种。
23.优选地,当所述表面活性剂为span-80和tween-60的复配液时,复配液中 span-80和tween-60的体积比为5~3:1。
24.进一步地,所述油相中表面活性剂的质量分数为1~8%。
25.进一步地,所述水相和油相的体积比为0.1~0.6:1。
26.进一步地,所述第一交联剂、第二交联剂均选自1,4-丁二醇二缩水甘油醚、戊二醛、edc、nhs、京尼平中的至少一种。
27.优选地,所述第一交联剂和第二交联剂采用edc和nhs的复配液,上述复配液中edc与nhs的摩尔比为5:1。
28.进一步地,本发明中,所述第一交联剂、第二交联剂的浓度为0.04%~1.5%;优选地,所述第二交联剂的浓度为0.05%~0.35%。
29.进一步地,所述缓冲溶液为磷酸盐缓冲溶液;优选地,所述磷酸盐缓冲溶液的ph值为5.0~7.0。
30.进一步地,采用ph 7.0~7.5的磷酸盐缓冲溶液作为透析液透析去除第一交联剂
和第二交联剂。
31.本发明采用上述透析液是为了去除第二交联剂的同时去除可能残留的第一交联剂(第一交联剂在步骤s3中已去除),以确保去除体系中所有的交联剂。
32.第二方面,提供了一种重组胶原蛋白双相凝胶,所述重组胶原蛋白双相凝胶采用如第一方面所述的制备方法制备而成。
33.第三方面,提供了如第二方面所述的重组胶原蛋白双相凝胶在制备软组织填充材料中的应用。
34.现有技术中,制备胶原蛋白微球采用先乳化、后交联的方式,会存在交联剂难以穿过已经形成的微球的水油界面,无法到达微球内部起到交联作用,更多的交联剂只是在油相中形成独立液滴或者在微球间形成交联,造成微球内交联度低、微球间过度交联甚至导致板块状的问题。
35.与现有技术相比,本发明具有以下有益效果:
36.1、本发明通过同步乳化交联方式,在乳化过程中,同时将交联剂滴入正在乳化的体系中,部分乳滴将交联剂直接包裹入胶原蛋白乳液颗粒的内部,乳液颗粒内部交联度高,部分未包裹交联剂或包裹少量交联剂的乳滴内部交联度小,从而形成多交联度分布的乳液颗粒,通过一次乳化交联反应,同时制备不同交联度的胶原蛋白微球,使得体系中胶原蛋白微球的交联度分布不同,可以实现梯度降解。
37.2、重组胶原蛋白a微球与重组胶原蛋白b凝胶混合时,通过加入少量交联剂,使得体系中同时存在重组胶原蛋白b自交联、重组胶原蛋白a微球和重组胶原蛋白b凝胶共交联的多种交联结构,从而保证胶原蛋白双相凝胶在体内的短期、中期、长期降解;另一方面,作为分散相的重组胶原蛋白b的分子量大于胶原微球制备时的重组胶原蛋白a的分子量,进一步实现梯度降解、保证了双相凝胶再体内的存留时间、实现长效降解。
38.3、不同于提取的胶原蛋白需用酸或碱溶解,本发明采用的重组胶原蛋白为水溶性的,溶液ph可以根据交联反应条件、交联反应机理的不同随时调节,且易于分散为目标粒径的微球,微球粒径可控性高。
39.4、本发明以重组胶原蛋白为原料,不存在种属特性,无免疫原性;且重组胶原蛋白分子量可控,可以方便的根据要求选择适宜分子量的重组胶原蛋白原料。
40.本发明附加的方面和优点将在下面的描述中部分给出,这些将从下面的描述中变得明显,或通过本发明的实践了解到。
附图说明
41.此处所说明的附图用来提供对本发明的进一步理解,构成本技术的一部分,并不构成对本发明的不当限定,在附图中:
42.图1为实施例3提供的双相凝胶的透射电镜照片;
43.图2为实施例3提供的双相凝胶的平均粒径及粒径分布示意图;
44.图3为实施例1-3的双相凝胶与对比例1-3的双相凝胶的弹性模量测试结果示意图;
45.图4为实施例2-3的双相凝胶与对比例2-3的双相凝胶的体外降解测试结果示意图。
具体实施方式
46.为了更充分的理解本发明的技术内容,下面将结合附图和具体实施例对本发明作进一步介绍和说明;显然,以下所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例;基于本发明中的实施例,本领域技术人员在没有作出创造性劳动前提下所获得的所有其它实施例,都属于本发明保护的范围。
47.对于本领域的技术人员来说,通过阅读本说明书公开的内容,本发明的特征、有益效果和优点将变得显而易见。
48.除非另外指明,所有百分比、分数和比率都是按本发明组合物的总质量计算的。本文术语“质量含量”可用符号“%”表示。
49.本文中“包括”、“包含”、“含”、“含有”、“具有”或其它变体意在涵盖非封闭式包括,这些术语之间不作区分。术语“包含”是指可加入不影响最终结果的其它步骤和成分。术语“包含”还包括术语“由...组成”和“基本上由...组成”。本发明的组合物和方法/工艺可包含、由其组成和基本上由本文描述的必要元素和限制项以及本文描述的任一的附加的或任选的成分、组分、步骤或限制项组成。
50.本发明采用的重组胶原蛋白均为现有的重组胶原蛋白。
51.实施例1
52.本实施例提供一种重组胶原蛋白双相凝胶,其包括重组胶原蛋白a微球和重组胶原蛋白b凝胶,重组胶原蛋白a微球均匀分散于重组胶原蛋白b凝胶中。
53.其中,重组胶原蛋白a微球的粒径为20~35μm,重组胶原蛋白a的分子量为30kda,重组胶原蛋白b的分子量为60kda。
54.上述重组胶原蛋白双相凝胶的制备方法,包括以下步骤:
55.s1:室温下,将重组胶原蛋白a(重组i型人源化胶原蛋白)溶解于ph 为7的磷酸盐缓冲溶液中,得到重组胶原蛋白a溶液,重组胶原蛋白a溶液的浓度为28mg/ml;
56.s2:以重组胶原蛋白a溶液为水相,加入到含有4%span-80的液体石蜡中,搅拌,水相和油相的体积比为0.3:1,同时边搅拌边滴加浓度为0.3%的戊二醛,于1000rpm均质乳化30min,继续搅拌4h,得到油包水乳状液;
57.s3:将步骤s2得到的油包水乳状液于5000rpm离心10min,弃去上层;再用石油醚、异丙醇、纯化水交替洗涤,每次清洗后于3000rpm离心5min收集微球,重复以上操3次;在40℃、-0.08mpa下真空干燥,得到重组胶原蛋白a微球;
58.s4:室温下,将重组胶原蛋白b(重组i型人源化胶原蛋白)溶解于ph 为7的磷酸盐缓冲溶液中,搅拌得到重组胶原蛋白b凝胶,重组胶原蛋白b凝胶的浓度为200mg/ml;将重组胶原蛋白a微球均匀分散于重组胶原蛋白b凝胶,调节重组胶原蛋白a微球的质量百分含量为60%,即得重组胶原蛋白双相凝胶。
59.实施例2
60.本实施例提供一种重组胶原蛋白双相凝胶,其包括重组胶原蛋白a微球和重组胶原蛋白b凝胶,重组胶原蛋白a微球均匀分散于重组胶原蛋白b凝胶中;重组胶原蛋白双相凝胶中重组胶原蛋白a微球的质量百分含量为40%。
61.其中,重组胶原蛋白a微球的粒径为40~60μm,重组胶原蛋白a的分子量为60kda,重组胶原蛋白b的分子量为90kda。
62.上述重组胶原蛋白双相凝胶的制备方法,包括以下步骤:
63.s1:室温下,将重组胶原蛋白a(重组i型类人胶原蛋白)溶解于ph为 7.0的磷酸盐缓冲溶液中,得到重组胶原蛋白a溶液,重组胶原蛋白a溶液的浓度为65mg/ml;
64.s2:以重组胶原蛋白a溶液为水相,加入到含有2%表面活性剂的医用白油中,表面活性剂为比例为4:1的span-80和tween-60的复配液,搅拌,水相和油相的体积比为0.25:1,边搅拌边滴加浓度为0.15%的戊二醛,于2000rpm 乳化45min,继续搅拌12h,得到油包水乳状液;
65.s3:将步骤s2得到的油包水乳状液于10000rpm离心10min,弃去上层;再用石油醚、异丙醇、纯化水交替洗涤,每次清洗后于5000rpm离心5min收集微球,重复以上操5次;在40℃、-0.08mpa下真空干燥,得到重组胶原蛋白a微球;
66.s4:室温下,将重组胶原蛋白b(重组i型类人胶原蛋白)溶解于ph为 5.5的磷酸盐缓冲溶液中,搅拌得到重组胶原蛋白b,重组胶原蛋白b凝胶的浓度为65mg/ml;
67.s5:将重组胶原蛋白a微球加入到重组胶原蛋白b凝胶中,边搅拌边滴加edc/nhs,其中,edc与nhs的摩尔比为5:1,edc/nhs浓度为0.35% (以edc记),在转速400rpm下交联2h,使重组胶原蛋白a微球均匀分散于重组胶原蛋白b凝胶中,然后放入截留分子量为8000~14000透析袋中透析,透析液为ph 7.0~7.5的磷酸盐缓冲溶液,去除残留交联剂;再置于40℃脱水至理论质量,即得重组胶原蛋白双相凝胶。
68.实施例3
69.本实施例提供一种重组胶原蛋白双相凝胶,其包括重组胶原蛋白a微球和重组胶原蛋白b凝胶,重组胶原蛋白a微球均匀分散于重组胶原蛋白b凝胶中;重组胶原蛋白双相凝胶中重组胶原蛋白a微球的质量百分含量为25%。
70.其中,重组胶原蛋白a微球的粒径为60~75μm,重组胶原蛋白a的分子量为100kda,重组胶原蛋白b的分子量为150kda。
71.上述重组胶原蛋白双相凝胶的制备方法,包括以下步骤:
72.s1:室温下,重组胶原蛋白a(重组iii型人源化胶原蛋白)溶解于ph为 5.5的磷酸盐缓冲溶液中,得到重组胶原蛋白a溶液;其中,重组胶原蛋白a 溶液的浓度为120mg/ml;
73.s2:以重组胶原蛋白a溶液为水相,加入到含有4%abil em90的液体石蜡中,搅拌,水相和油相的体积比为0.4:1,边搅拌边滴加交联剂edc/nhs;其中,edc与nhs的摩尔比为5:1,edc/nhs浓度为1.5%(以edc记),于转速2200rpm下乳化40min,继续搅拌12h,得到油包水乳状液;
74.s3:将步骤s2得到的乳状液于12000rpm离心10min,弃去上层;再用石油醚、异丙醇、纯化水交替洗涤,每次清洗后于6000rpm离心5min收集微球,重复以上操4次;在40℃、-0.08mpa下真空干燥,得到重组胶原蛋白a微球;
75.s4:室温下,将重组胶原蛋白b(重组iii型人源化胶原蛋白)溶解于ph 为5.5的磷酸盐缓冲溶液中,搅拌得到重组胶原蛋白b凝胶,重组胶原蛋白b 凝胶的浓度为130mg/ml;
76.s5:将重组胶原蛋白a微球加入到重组胶原蛋白b凝胶中,边搅拌边滴加edc/nhs,其中,edc与nhs的摩尔比为5:1,edc/nhs浓度为0.15% (以edc记),在转速600rpm下交联2h,使重组胶原蛋白a微球均匀分散于重组胶原蛋白b凝胶中,然后放入截留分子量为8000~14000透析袋中透析,透析液为ph 7.0~7.5的磷酸盐缓冲溶液,去除残留交联剂;再置于
40℃脱水至理论质量,即得重组胶原蛋白双相凝胶。
77.对比例1
78.对比例1的制备方法与实施例1相比,区别仅在于:
79.步骤s2中,采用先乳化获得乳状液后再交联的方式,制备得到重组胶原蛋白双相凝胶。
80.对比例2
81.对比例2的制备方法与实施例2相比,区别在于:
82.步骤s2中,采用先乳化获得乳状液后再交联的方式,步骤s5中,在重组胶原蛋白a微球和b凝胶混合的过程中不加入交联剂,制备得到重组胶原蛋白双相凝胶。
83.本对比例的重组胶原蛋白双相凝胶,其包括重组胶原蛋白a微球和重组胶原蛋白b凝胶,重组胶原蛋白a微球均匀分散于重组胶原蛋白b凝胶中;重组胶原蛋白双相凝胶中双相凝胶a的质量百分含量为25%。
84.其中,重组胶原蛋白a微球的粒径为50~75μm,重组胶原蛋白a的分子量为90kda,重组胶原蛋白b的分子量为90kda。
85.上述重组胶原蛋白双相凝胶的制备方法,包括以下步骤:
86.s1:室温下,重组胶原蛋白a(重组i型类人胶原蛋白)溶解于ph为7 的磷酸盐缓冲溶液中,得到重组胶原蛋白a溶液;其中,重组胶原蛋白a溶液的浓度为65mg/ml;
87.s2:以重组胶原蛋白a溶液为水相,加入到含有2%体积比为4:1的 span-80和tween-60的复配液的硅油中,搅拌,水相和油相的体积比为0.2:1,于1200rpm搅拌1h,得到乳状液体;
88.s3:在搅拌状态下(1200rpm)向s2所得乳状液体中缓慢加入浓度为0.5%的戊二醛,交联2h;
89.s4:将步骤s3得到的乳状液于10000rpm离心10min,弃去上层;再用石油醚、异丙醇、纯化水交替洗涤,每次清洗后于5000rpm离心5min收集微球,重复以上操3次;40℃、-0.08mpa真空干燥,得到重组胶原蛋白a微球;
90.s5:室温下,将重组胶原蛋白b(重组i型类人胶原蛋白)溶解于ph为 7.0的磷酸盐缓冲溶液中,搅拌得到重组胶原蛋白b凝胶,重组胶原蛋白b凝胶的浓度为65mg/ml,将重组胶原蛋白a微球分散于重组胶原蛋白b凝胶,即得重组胶原蛋白双相凝胶。
91.对比例3
92.对比例3的制备方法与实施例1相比,区别在于:
93.步骤s2中采用同步乳化交联的方式,获得重组胶原蛋白a微球;在重组胶原蛋白a微球和b凝胶混合的过程中不加入交联剂,制备得到重组胶原蛋白双相凝胶。
94.本对比例的重组胶原蛋白双相凝胶,其包括重组胶原蛋白a微球和重组胶原蛋白b凝胶,重组胶原蛋白a微球均匀分散于重组胶原蛋白b凝胶中;重组胶原蛋白双相凝胶中双相凝胶a的质量百分含量为40%。
95.其中,重组胶原蛋白a微球的粒径为50~65μm,重组胶原蛋白a的分子量为60kda,重组胶原蛋白b的分子量为90kda。
96.上述重组胶原蛋白双相凝胶的制备方法,包括以下步骤:
97.s1:室温下,重组胶原蛋白a(重组iii型人源化胶原蛋白)溶解于ph为 5.5的磷酸
盐缓冲溶液中,得到重组胶原蛋白a溶液;其中,重组胶原蛋白a 溶液的浓度为50mg/ml;
98.s2:以重组胶原蛋白a溶液为水0.1:1,边搅拌边滴加交联剂edc/nhs;其中,edc与nhs的摩尔比为5:1,edc/nhs浓度为1.5%(以edc记),于转速1000rpm下乳化35min,继续搅拌6h,得到油包水乳状液;
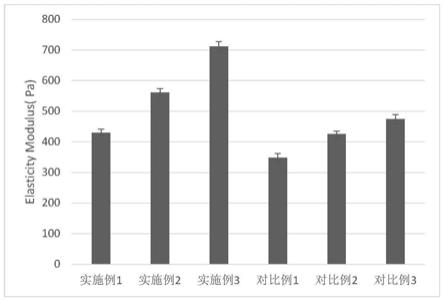
99.s3:将s2得到的油包水乳状液于10000rpm离心10min,弃去上层;再用石油醚、异丙醇、纯化水交替洗涤,每次清洗后于5000rpm离心5min收集微球,重复以上操3次;在40℃、-0.08mpa下真空干燥,得到重组胶原蛋白a 微球;
100.s4:室温下,将重组胶原蛋白b(重组iii型人源化胶原蛋白)溶解于ph 为7.0的磷酸盐缓冲溶液中,搅拌得到重组胶原蛋白b凝胶;其中,重组胶原蛋白b凝胶的浓度为100mg/ml,;
101.s5:将重组胶原蛋白a微球加入到重组胶原蛋白b凝胶中,搅拌,使重组胶原蛋白a微球均匀分散于重组胶原蛋白b凝胶中,脱水,即得重组胶原蛋白双相凝胶。
102.效果测试
103.微球外观
104.对实施例3制备得到的双相凝胶进行透射电镜照片,其电镜照片如图1所示,实施例3的平均粒径及粒径分布示意图如图2所示。
105.弹性模量测试
106.将本发明实施例1-3制备的双相凝胶与对比例1-3制备的凝胶进行弹性模量测试,其结果如图3所示。
107.根据图3的测试结果可得,采用本发明技术方案制备的双相凝胶,其弹性模量普遍高于对比例;具体而言,与对比例相较,采用的同步乳化交联方式制备的胶原蛋白微球双相凝胶,其弹性模量明显高于先乳化、后交联的对比例1 和对比例2;实施例2和实施例3采用的同步乳化交联以及二次交联的方式,显著提高了双相凝胶体系的弹性模量,说明采用本发明技术方案获得的双相凝胶微球的网络结构致密,具有较高的抵抗变形的能力。
108.体外降解测试
109.采用0.5mg/ml胶原酶对实施例2-3、对比例2-3的双相凝胶微球进行体外降解。在1、2、3、4、5、6周后,分别计算其质量损失率。
110.具体测试过程为:将干燥的无菌双相凝胶浸泡在含4ml酶溶液的试管中,试管放置于37.0℃恒温箱中,浸泡1、2、3、4、5、6周后,从酶溶液中取出样品,冻干并称重。重量损失按w
l
=(w
0-w1)/w0*100%计算,其中w0和w1分别代表样品胶原酶溶液浸泡前后的样品的质量,测试结果如图4所示。
111.根据图4的测试结果可知,在测试的降解周期内,实施例组的质量损失率普遍低于对比例组,采用同步乳化交联以及二次交联的方式进行制备,能显著提升双相凝胶体系的交联密度,体系中同时存在多种交联方式,包括微球内部交联、微球间交联及微球与凝胶基质的交联,其中,同步乳化过程中交联剂同步被包裹入微球内部,减少了先乳化、后交联时交联剂穿透水油界面的阻力,能够在微球内部发生高效交联;另一方面,凝胶基质的分子量筛选以及适度的交联,既保证了双相凝胶适宜的弹性模量和可推注性,又与微球形成梯度交联,保证了产品使用后的中、长期效果。
112.体内植入实验
113.选取6只新西兰兔,雌雄不限,体重1.7~2kg,日龄70~90d,在每只兔耳背部选择4个注射区域,分别注射实施例3和对照例3制备的双相凝胶0.1ml,每个样品注射2个平行区域,采用三维光学形状轮廓测量仪,以皮肤隆起的边缘外4mm的组织为界,切取包括软骨在内的兔耳全层,对标本进行扫描及建模,于注射后、1w、4w、12w、32w、52w,分别取材测量,评价体内植入后的体积增量,结果如表1所示。
114.表1:
115.体积增量实施例3对比例3注射后(mm3)131.664
±
14.784126.785
±
12.5681w(mm3)153.651
±
15.742132.686
±
10.3574w(mm3)162.631
±
16.544103.158
±
13.54512w(mm3)148.597
±
14.36677.856
±
15.22732w(mm3)117.688
±
10.78165.694
±
17.44852w(mm3)121.152
±
12.76151.331
±
16.552
116.根据表1的测量结果可得,采用本发明同步乳化交联以及二次交联的方式制备的双相凝胶,可以实现缓慢梯度降解,在填充6~8个月之后,作为基质的重组胶原蛋白b凝胶发生降解,之后主要是交联度更高的微球在体内的体积支撑和缓慢降解,并刺激胶原再生,实现了中期、长期体积增量,填充效果更加自然;而对比例3的双相凝胶未经二次交联,重组胶原蛋白b凝胶降解相对较快,在1~3月时即发生明显降解,难以保证中、长期填充效果。
117.以上对本发明实施例所提供的技术方案进行了详细介绍,本文中应用了具体个例对本发明实施例的原理以及实施方式进行了阐述,以上实施例的说明只适用于帮助理解本发明实施例的原理;同时,对于本领域的一般技术人员,依据本发明实施例,在具体实施方式以及应用范围上均会有改变之处,综上所述,本说明书内容不应理解为对本发明的限制。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1