一种以湿料合成对二亚硝基苯的制备方法与流程
1.本发明属于化工原材料生产领域,尤其涉及一种化工原料对二亚硝基苯的制备方法。
背景技术:
2.对二亚硝基苯(1,4-dinitrosobenzene)是一种具有棕色粉末或固体外观的硫化剂,对二亚硝基苯的别名为1,4-对二亚硝基苯,分子量为136.11,熔点为146.5℃,密度为1.4175,主要用于丁基胶,天然胶,丁苯胶等胶体的硫化粘接。
3.橡胶-金属复合件集橡胶和金属的特性于一身,在汽车、机械、国防等行业得到了越来越广泛的应用,而橡胶-金属间牢固的粘接是其使用可靠性的关键。随着科技的进步和经济的发展,合成胶粘剂正在越来越多地代替机械联结,胶粘剂的应用已扩展到许多领域,为各行各业简化工艺、节约能源、降低成本、提高经济效益提供了一条有效的途径,并已成为一个极具发展前景的精细化工行业。除工艺无法操作或基材在热硫化条件下不稳定外,橡胶-金属的粘接均采用硫化粘接的方式。因此用于硫化粘接的胶粘剂得到了广泛研究,新的胶粘剂不断涌现。多亚硝基芳香类化合物特别是对二亚硝基苯因其优异的粘接促进作用在这些胶粘剂中得到广泛使用。而对二亚硝基苯的产品质量直接影响到其硫化胶粘性能,高品质的对二亚硝基苯可以有效提高对橡胶的胶粘效果。
4.对二硝基苯可能以聚合物或伴生形式存在。因此,对二亚硝基苯的化学反应活性会随着样品制备方式的不同而变化。例如,ruggli和petitjean制备的对二亚硝基苯样品,这些样品几乎与苯胺可以发生定量反应,而其他样品则完全没有反应。在对二亚硝基苯硫化橡胶类材料反应性方面也发现了类似的差异。高反应性的对二亚硝基苯在弹性体合成橡胶的硫化中产生的效果,需要相对数量更大的低反应性对二亚硝基苯才能做到。在相同的基本分析条件下,活性样品和非活性样品类似的差异不能用简单的分析判断来解释,产品的实际化学差异尚不清楚,但可能是由于结构和聚合程度的不同而导致反应性的差异。
5.关于对二亚硝基苯的制备,早期是在碱性溶液中使用铁氰化钾氧化制备得到,但由于铁氰化钾分子量大,氧化一份对苯醌二肟需要两份铁氰化钾,因此需要较大量的铁氰化钾进行这个反应。除了成本外,该工艺还存在其他缺点,特别是当产品用于硫化弹性合成橡胶材料时,去除反应中形成的大量亚铁氰化物非常困难,因为对二硝基苯以一种紧密分裂的状态下降,因此亚铁氰化物被封闭,需要大量的水和相当长的时间来去除这种杂质。除去亚铁氰化物是很重要的步骤,因为像其他离子键材料一样,它对聚合物硫化胶的电绝缘性能具有不良的影响。而且亚铁氰化物是一种氧化催化剂,会显著降低聚合物的耐老化性能。
6.目前,合成对二亚硝基苯最为实用的方式是美国专利us.2419976提到的,通过碱金属的次氯酸盐或次溴酸盐氧化对苯醌二肟的碱性溶液,该反应在相对较低的温度下进行,因此没有大量的二亚硝基化合物被氧化成二硝基。用次卤酸盐代替铁氰化钾可以减少氧化剂的用量以及产物中的电解质,并获得收率优异的对二亚硝基苯,这种方式合成的对
二亚硝基苯在合成橡胶材料的硫化中具有高反应性。但是,作为合成对二亚硝基苯的工业原料对苯醌二肟,其成本高,施工操作时干粉粉尘很大,伴随着粉尘污染、粉尘爆炸等潜在问题有待解决。
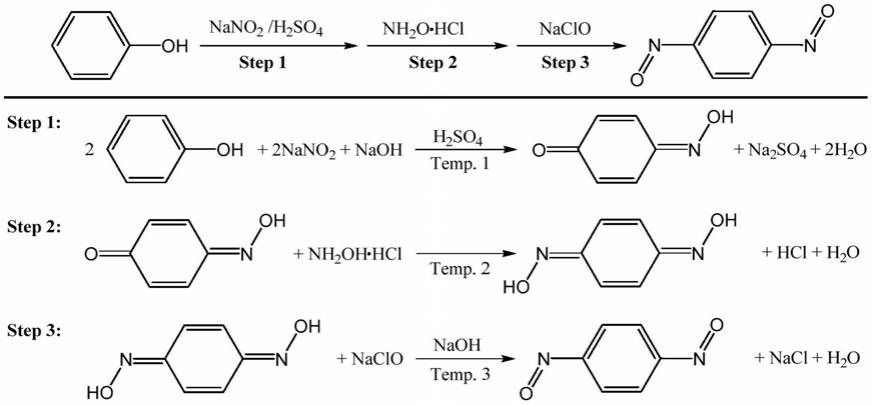
技术实现要素:
7.本发明实施例的目的是提供一种以湿料合成对二亚硝基苯的方法,解决了传统技术中副产物难以除净导致的橡胶材质质量降低,或合成工艺复杂、原料成本高、原料使用存在潜在危险等问题。
8.根据本技术实施例,提供一种以湿料合成对二亚硝基苯的制备方法,包括:
9.(1)将苯酚加入亚硝酸钠、无机碱的水溶液,配成苯酚钠、亚硝酸钠的混合溶液,在预定温度下,将混合溶液滴加到硫酸溶液中,搅拌反应,过滤、洗涤制得中间体对亚硝基苯酚湿料;
10.(2)在预定温度下,将盐酸羟胺水溶液滴加到中间体对亚硝基苯酚湿料中,搅拌反应,过滤、洗涤制得对苯醌二肟湿料;
11.(3)将对苯醌二肟湿料溶解于无机碱的水溶液中,得到碱性对苯醌二肟溶液,在预定温度及反应过程环境完全避光处理情况下,将有效氯为5%-10%naclo稀溶液滴加到碱性对苯醌二肟溶液中,搅拌反应,过滤、洗涤制得对二亚硝基苯产物。
12.本技术上述制备对二亚硝基苯的方法,其结构式如下:
[0013][0014]
优选地,在(1)、(2)中,所述苯酚、亚硝酸钠、无机碱、硫酸、盐酸羟胺的摩尔比为1.0:(1.0-2.0):(1.0-1.5):(1.0-3.0):(1.0-2.0)。
[0015]
优选地,在(1)中,预定温度为-25~5℃,滴加的时长为1-5小时。
[0016]
优选地,在(1)中,洗涤过程的洗涤效果达到所出滤液测得的ph值为中性。
[0017]
优选地,在(2)中,预定温度为40~70℃。
[0018]
优选地,在(3)中,对苯醌二肟湿料中的苯醌二肟、无机碱、水的质量比为1:0.53:12.5。
[0019]
优选地,在(3)中,滴加时长为0.5-2小时。
[0020]
优选地,在(3)中,对苯醌二肟湿料中的苯醌二肟跟次氯酸钠的摩尔比为1.0:(0.8-1.5)。
[0021]
优选地,在(3)中,预定温度为5~25℃,滴加时长为1-4小时。
[0022]
优选地,在(3)中,洗涤过程的洗涤效果达到所出滤液测得的ph值为中性。
[0023]
该方法从最基础的底物苯酚出发,三步反应连续不中断,合成过程中所涉及的中间体湿料均无需干燥,直接用于对二亚硝基苯的合成。
[0024]
需要说明的是,所述的合成过程中,三步反应连续不中断,所涉及的中间体湿料均无需干燥。反应过程中,混合溶液溶液、盐酸羟胺水溶液、naclo稀溶液的滴加均采用连续匀速加入。反应过程中,湿料对苯醌二肟的固含量通过测试水分进而计算得出。反应过程中,naclo溶液的有效氯测试值保质期为2天。
[0025]
本技术的实施例提供的技术方案可以包括以下有益效果:
[0026]
由上述实施例可知,本技术的方法原料从苯酚出发,经过三步连续釜式反应最终得到对二亚硝基苯产品,此法原料成本低、全过程有仅涉及到水作为反应溶剂、操作工艺简单、过程中间产物湿料不需要干燥,工艺简化,最终制得的对二亚硝基苯具有优异的胶黏粘接性能。该方法适合于对二亚硝基苯产品的工业化生产或连续化扩量生产,具有很好的应用性前景。其中,反应过程中间体对亚硝基苯酚、对苯醌二肟亦可烘干直接作为化工产品用于销售(对苯醌二肟目前也是市场上应用广泛的热硫化剂产品之一)。
[0027]
应当理解的是,以上的一般描述和后文的细节描述仅是示例性和解释性的,并不能限制本技术。
附图说明
[0028]
图1为实施例1-实施例8所得的对二亚硝基苯样品的红外谱图,其中各实施例纵坐标相互独立。
[0029]
图2为图1的局部放大图,其中红外谱的纵坐标在统一标准下。
[0030]
图3为实施例9-实施例17所得的对二亚硝基苯样品的红外谱图,其中各实施例纵坐标相互独立。
[0031]
图4为图3的局部放大图,其中红外谱的纵坐标在统一标准下。
[0032]
图5为实施例1和对比例1所得的对二亚硝基苯样品的红外谱图,其中各实施例纵坐标相互独立。
[0033]
图6为图5的局部放大图,其中红外谱的纵坐标在统一标准下。
具体实施方式
[0034]
这里将详细地对示例性实施例进行说明,以下示例性实施例中所描述的实施方式并不代表与本技术相一致的所有实施方式。
[0035]
在本技术使用的术语是仅仅出于描述特定实施例的目的,而非旨在限制本技术。在本技术和所附权利要求书中所使用的单数形式的“一种”、“所述”和“该”也旨在包括多数形式,除非上下文清楚地表示其他含义。还应当理解,本文中使用的术语“和/或”是指并包含一个或多个相关联的列出项目的任何或所有可能组合。
[0036]
实施例1:
[0037]
第一步,将亚硝酸钠(2.0mol,137.99g)和氢氧化钠(1.5mol,60.12g)加入483ml自来水,搅拌使之完全溶解,再加入苯酚(1.0mol,94.60g),搅拌溶解制得混合溶液1;将浓硫
酸(3mol,294.78g)缓慢加入535ml冰水中,搅拌稀释制得溶液2;
[0038]
在3℃的环境温度下,将溶液1缓慢(2小时)滴加到溶液2中,维持体系温度在5℃,加毕再持续搅拌2小时,过滤、洗涤制得中间体对亚硝基苯酚湿料;
[0039]
第二步,将盐酸羟胺(2mol,139.54g)加入109ml自来水,搅拌溶解制得溶液3;在42℃的环境温度下,将溶液3(2小时)滴加到中间体对亚硝基苯酚湿料的水溶液中,维持体系温度在40℃,加毕再持续搅拌2小时,过滤、洗涤制得对苯醌二肟湿料(湿重:132.09)。取样(5.1203g)置于鼓风烘箱,烘干至恒重(3.4429g),测得样品固含量:67.24%,对苯醌二肟产率:60.5%;
[0040]
第三步,将对苯醌二肟湿料(0.618mol,126.97g,湿料)溶解于氢氧化钠(1.12mol,44.96g)的水溶液(970ml)中,配置成碱性对苯醌二肟溶液4;将naclo溶液(有效氯8.62%,0.680mol,559.54g)加入405ml自来水,配置成有效氯为5%naclo溶液稀溶液5;在13℃的环境温度及反应容器完全避光处理的条件下,将溶液5(4小时)滴加到溶液4中,维持体系温度在15℃,加毕,自然温度再持续搅拌2小时,过滤、打浆洗涤、烘干制得对二亚硝基苯产物,干重:69.23g,产率:82.30%(以对苯醌二肟为原料计算),灼烧残渣:0.52%。
[0041]
本实施例中在第三步反应合成前,对naclo溶液的有效氯进行测定,有效氯数值两天内有效;
[0042]
本实施例中的第一步和第三步的反应中的naoh也可以是koh、nahco3、na2co3、khco3、k2co3等无机碱中的任何一种替代。
[0043]
实施例2:
[0044]
第一步,将亚硝酸钠(1.0mol,69.21g)和氢氧化钠(1.5mol,60.23g)加入483ml自来水,搅拌使之完全溶解,再加入苯酚(1.0mol,94.52g),搅拌溶解制得混合溶液1;将浓硫酸(3mol,294.03g)缓慢加入535ml冰水中,搅拌稀释制得溶液2;
[0045]
在3℃的环境温度下,将溶液1缓慢(2小时)滴加到溶液2中,维持体系温度在5℃,加毕再持续搅拌2小时,过滤、洗涤制得中间体对亚硝基苯酚湿料;
[0046]
第二步,将盐酸羟胺(2mol,139.02g)加入109ml自来水,搅拌溶解制得溶液3;在42℃的环境温度下,将溶液3(2小时)滴加到中间体对亚硝基苯酚湿料的水溶液中,维持体系温度在40℃,加毕再持续搅拌2小时,过滤、洗涤制得对苯醌二肟湿料(湿重:112.76)。取样(5.0022g)置于鼓风烘箱,烘干至恒重(3.7071g),测得样品固含量:74.11%,对苯醌二肟产率:60.5%;
[0047]
第三步,将对苯醌二肟湿料(0.578mol,107.76g,湿料)溶解于氢氧化钠(1.05mol,42.06g)的水溶液(970ml)中,配置成碱性对苯醌二肟溶液4;将naclo溶液(有效氯8.62%,0.636mol,523.42g)加入379ml自来水,配置成有效氯为5%naclo溶液稀溶液5;在13℃的环境温度及反应容器完全避光处理的条件下,将溶液5(4小时)滴加到溶液4中,维持体系温度在15℃,加毕,自然温度再持续搅拌2小时,过滤、打浆洗涤、烘干制得对二亚硝基苯产物,干重:63.86g,产率:81.17%(以对苯醌二肟为原料计算)。灼烧残渣:0.28%。
[0048]
本实施例中第一步底物亚硝酸钠的配比为1.5个当量,也可以是1.0个当量或是1.0-2.0个当量之间的任意当量;
[0049]
本实施例中在第三步反应合成前,对naclo溶液的有效氯进行测定,有效氯数值两天内有效;
[0050]
本实施例中的第一步和第三步的反应中的naoh也可以是koh、nahco3、na2co3、khco3、k2co3中的任何一种替代。
[0051]
实施例3:
[0052]
第一步,将亚硝酸钠(2.0mol,138.11g)和氢氧化钠(1.0mol,40.34g)加入483ml自来水,搅拌使之完全溶解,再加入苯酚(1.0mol,94.42g),搅拌溶解制得混合溶液1;将浓硫酸(3mol,294.12g)缓慢加入535ml冰水中,搅拌稀释制得溶液2;
[0053]
在3℃的环境温度下,将溶液1缓慢(2小时)滴加到溶液2中,维持体系温度在5℃,加毕再持续搅拌2小时,过滤、洗涤制得中间体对亚硝基苯酚湿料;
[0054]
第二步,将盐酸羟胺(2mol,139.24g)加入109ml自来水,搅拌溶解制得溶液3;在42℃的环境温度下,将溶液3(2小时)滴加到中间体对亚硝基苯酚湿料的水溶液中,维持体系温度在40℃,加毕再持续搅拌2小时,过滤、洗涤制得对苯醌二肟湿料(湿重:120.59g)。取样(5.0045g)置于鼓风烘箱,烘干至恒重(3.5082g),测得样品固含量:70.10%,对苯醌二肟产率:61.20%;
[0055]
第三步,将对苯醌二肟湿料(0.587mol,115.59g,湿料)溶解于氢氧化钠(1.07mol,42.67g)的水溶液(1013ml)中,配置成碱性对苯醌二肟溶液4;将naclo溶液(有效氯8.54%,0.645mol,536.05g)加入380ml自来水,配置成有效氯为5%naclo溶液稀溶液5;在13℃的环境温度及反应容器完全避光处理的条件下,将溶液5(4小时)滴加到溶液4中,维持体系温度在15℃,加毕,自然温度再持续搅拌2小时,过滤、打浆洗涤、烘干制得对二亚硝基苯产物,干重:63.95g,产率:80.10%(以对苯醌二肟为原料计算),灼烧残渣:0.24%。
[0056]
本实施例中第一步底物氢氧化钠的配比为1.0个当量,也可以是1.2个当量或是1.0-1.5个当量之间的任意当量;
[0057]
本实施例中在第三步反应合成前,对naclo溶液的有效氯进行测定,有效氯数值两天内有效;
[0058]
本实施例中的第一步和第三步的反应中的naoh也可以是koh、nahco3、na2co3、khco3、k2co3中的任何一种替代。
[0059]
实施例4:
[0060]
第一步,将亚硝酸钠(2.0mol,138.45g)和氢氧化钠(1.5mol,60.02g)加入483ml自来水,搅拌使之完全溶解,再加入苯酚(1.0mol,94.15g),搅拌溶解制得混合溶液1;将浓硫酸(1mol,98.17g)缓慢加入535ml冰水中,搅拌稀释制得溶液2;
[0061]
在3℃的环境温度下,将溶液1缓慢(2小时)滴加到溶液2中,维持体系温度在5℃,加毕再持续搅拌2小时,过滤、洗涤制得中间体对亚硝基苯酚湿料;
[0062]
第二步,将盐酸羟胺(2mol,138.99g)加入109ml自来水,搅拌溶解制得溶液3;在42℃的环境温度下,将溶液3(2小时)滴加到中间体对亚硝基苯酚湿料的水溶液中,维持体系温度在40℃,加毕再持续搅拌2小时,过滤、洗涤制得对苯醌二肟湿料(湿重:105.75g)。取样(5.2051g)置于鼓风烘箱,烘干至恒重(4.0142g),测得样品固含量:77.12%,对苯醌二肟产率:59.04%;
[0063]
第三步,将对苯醌二肟湿料(0.561mol,100.54g,湿料)溶解于氢氧化钠(1.02mol,40.84g)的水溶液(946ml)中,配置成碱性对苯醌二肟溶液4;将naclo溶液(有效氯8.12%,0.617mol,539.50g)加入337ml自来水,配置成有效氯为5%naclo溶液稀溶液5;在13℃的环
境温度及反应容器完全避光处理的条件下,将溶液5(4小时)滴加到溶液4中,维持体系温度在15℃,加毕,自然温度再持续搅拌2小时,过滤、打浆洗涤、烘干制得对二亚硝基苯产物,干重:62.04g,产率:78.02%(以对苯醌二肟为原料计算),灼烧残渣:0.62%。
[0064]
本实施例中第一步底物浓硫酸的配比为2.0个当量,也可以是1.0个当量或是1.0-3.0个当量之间的任意当量;
[0065]
本实施例中在第三步反应合成前,对naclo溶液的有效氯进行测定,有效氯数值两天内有效;
[0066]
本实施例中的第一步和第三步的反应中的naoh也可以是koh、nahco3、na2co3、khco3、k2co3中的任何一种替代。
[0067]
实施例5:
[0068]
第一步,将亚硝酸钠(2.0mol,138.12g)和氢氧化钠(1.5mol,60.14g)加入483ml自来水,搅拌使之完全溶解,再加入苯酚(1.0mol,94.13g),搅拌溶解制得混合溶液1;将浓硫酸(3.0mol,295.31g)缓慢加入535ml冰水中,搅拌稀释制得溶液2;
[0069]
在3℃的环境温度下,将溶液1缓慢(2小时)滴加到溶液2中,维持体系温度在5℃,加毕再持续搅拌2小时,过滤、洗涤制得中间体对亚硝基苯酚湿料;
[0070]
第二步,将盐酸羟胺(1.0mol,169.57g)加入109ml自来水,搅拌溶解制得溶液3;在42℃的环境温度下,将溶液3(2小时)滴加到中间体对亚硝基苯酚湿料的水溶液中,维持体系温度在40℃,加毕再持续搅拌2小时,过滤、洗涤制得对苯醌二肟湿料(湿重:123.37g)。取样(4.8920g)置于鼓风烘箱,烘干至恒重(3.3838g),测得样品固含量:69.17%,对苯醌二肟产率:61.78%;
[0071]
第三步,将对苯醌二肟湿料(0.593mol,118.48g,湿料)溶解于氢氧化钠(1.08mol,43.16g)的水溶液(987.88ml)中,配置成碱性对苯醌二肟溶液4;将naclo溶液(有效氯7.87%,0.653mol,588.33g)加入338ml自来水,配置成有效氯为5%naclo溶液稀溶液5;在13℃的环境温度及反应容器完全避光处理的条件下,将溶液5(4小时)滴加到溶液4中,维持体系温度在15℃,加毕,自然温度再持续搅拌2小时,过滤、打浆洗涤、烘干制得对二亚硝基苯产物,干重:66.04g,产率:81.78%(以对苯醌二肟为原料计算),灼烧残渣:0.64%。
[0072]
本实施例中第二步底物盐酸羟胺的配比为1.5个当量,也可以是1.0个当量或是1.0-2.0个当量之间的任意当量;
[0073]
本实施例中在第三步反应合成前,对naclo溶液的有效氯进行测定,有效氯数值两天内有效;
[0074]
本实施例中的第一步和第三步的反应中的naoh也可以是koh、nahco3、na2co3、khco3、k2co3中的任何一种替代。
[0075]
实施例6:
[0076]
第一步,将亚硝酸钠(2.0mol,138.15g)和氢氧化钠(1.5mol,60.21g)加入483ml自来水,搅拌使之完全溶解,再加入苯酚(1.0mol,94.42g),搅拌溶解制得混合溶液1;将浓硫酸(3.0mol,294.14g)缓慢加入535ml冰水中,搅拌稀释制得溶液2;
[0077]
在-12℃的环境温度下,将溶液1缓慢(2小时)滴加到溶液2中,维持体系温度在-10℃,加毕再持续搅拌2小时,过滤、洗涤制得中间体对亚硝基苯酚湿料;
[0078]
第二步,将盐酸羟胺(2.0mol,138.99g)加入109ml自来水,搅拌溶解制得溶液3;在
42℃的环境温度下,将溶液3(2小时)滴加到中间体对亚硝基苯酚湿料的水溶液中,维持体系温度在40℃,加毕再持续搅拌2小时,过滤、洗涤制得对苯醌二肟湿料(湿重:118.04g)。取样(5.3020g)置于鼓风烘箱,烘干至恒重(3.9341g),测得样品固含量:74.20%,对苯醌二肟产率:63.41%;
[0079]
第三步,将对苯醌二肟湿料(0.606mol,112.74g,湿料)溶解于氢氧化钠(1.10mol,44.06g)的水溶液(1017ml)中,配置成碱性对苯醌二肟溶液4;将naclo溶液(有效氯7.61%,0.666mol,621.06g)加入324ml自来水,配置成有效氯为5%naclo溶液稀溶液5;在13℃的环境温度及反应容器完全避光处理的条件下,将溶液5(4小时)滴加到溶液4中,维持体系温度在15℃,加毕,自然温度再持续搅拌2小时,过滤、打浆洗涤、烘干制得对二亚硝基苯产物,干重:67.60g,产率:82.01%(以对苯醌二肟为原料计算),灼烧残渣:0.50%。
[0080]
本实施例中第一步反应温度为-10℃,也可以是-25℃或是-25℃-5℃之间任意温度;
[0081]
本实施例中在第三步反应合成前,对naclo溶液的有效氯进行测定,有效氯数值两天内有效;
[0082]
本实施例中的第一步和第三步的反应中的naoh也可以是koh、nahco3、na2co3、khco3、k2co3中的任何一种替代。
[0083]
实施例7:
[0084]
第一步,将亚硝酸钠(2.0mol,138.10g)和氢氧化钠(1.5mol,60.27g)加入483ml自来水,搅拌使之完全溶解,再加入苯酚(1.0mol,94.07g),搅拌溶解制得混合溶液1;将浓硫酸(3.0mol,294.45g)缓慢加入535ml冰水中,搅拌稀释制得溶液2;
[0085]
在-27℃的环境温度下,将溶液1缓慢(2小时)滴加到溶液2中,维持体系温度在-25℃,加毕再持续搅拌2小时,过滤、洗涤制得中间体对亚硝基苯酚湿料;
[0086]
第二步,将盐酸羟胺(2.0mol,139.07g)加入109ml自来水,搅拌溶解制得溶液3;在42℃的环境温度下,将溶液3(2小时)滴加到中间体对亚硝基苯酚湿料的水溶液中,维持体系温度在40℃,加毕再持续搅拌2小时,过滤、洗涤制得对苯醌二肟湿料(湿重:121.21g)。取样(5.0301g)置于鼓风烘箱,烘干至恒重(3.4778g),测得样品固含量:69.14%,对苯醌二肟产率:60.67%;
[0087]
第三步,将对苯醌二肟湿料(0.582mol,116.18g,湿料)溶解于氢氧化钠(1.06mol,42.30g)的水溶液(968ml)中,配置成碱性对苯醌二肟溶液4;将naclo溶液(有效氯7.28%,0.666mol,623.38g)加入284ml自来水,配置成有效氯为5%naclo溶液稀溶液5;在13℃的环境温度及反应容器完全避光处理的条件下,将溶液5(4小时)滴加到溶液4中,维持体系温度在15℃,加毕,自然温度再持续搅拌2小时,过滤、打浆洗涤、烘干制得对二亚硝基苯产物,干重:63.17g,产率:79.81%(以对苯醌二肟为原料计算),灼烧残渣:0.45%。
[0088]
本实施例中第一步反应温度为-25℃,也可以是-25℃或是-25℃-5℃之间任意温度;
[0089]
本实施例中在第三步反应合成前,对naclo溶液的有效氯进行测定,有效氯数值两天内有效;
[0090]
本实施例中的第一步和第三步的反应中的naoh也可以是koh、nahco3、na2co3、khco3、k2co3中的任何一种替代。
[0091]
实施例8:
[0092]
第一步,将亚硝酸钠(2.0mol,138.08g)和氢氧化钠(1.5mol,60.19g)加入483ml自来水,搅拌使之完全溶解,再加入苯酚(1.0mol,94.15g),搅拌溶解制得混合溶液1;将浓硫酸(3.0mol,294.19g)缓慢加入535ml冰水中,搅拌稀释制得溶液2;
[0093]
在3℃的环境温度下,将溶液1缓慢(1小时)滴加到溶液2中,维持体系温度在5℃,加毕再持续搅拌2小时,过滤、洗涤制得中间体对亚硝基苯酚湿料;
[0094]
第二步,将盐酸羟胺(2.0mol,139.17g)加入109ml自来水,搅拌溶解制得溶液3;在42℃的环境温度下,将溶液3(2小时)滴加到中间体对亚硝基苯酚湿料的水溶液中,维持体系温度在40℃,加毕再持续搅拌2小时,过滤、洗涤制得对苯醌二肟湿料(湿重:127.12g)。取样(5.1012g)置于鼓风烘箱,烘干至恒重(3.4678g),测得样品固含量:67.98%,对苯醌二肟产率:62.56%;
[0095]
第三步,将对苯醌二肟湿料(0.600mol,122.02g,湿料)溶解于氢氧化钠(1.09mol,43.70g)的水溶液(998ml)中,配置成碱性对苯醌二肟溶液4;将naclo溶液(有效氯7.28%,0.661mol,643.72g)加入294ml自来水,配置成有效氯为5%naclo溶液稀溶液5;在13℃的环境温度及反应容器完全避光处理的条件下,将溶液5(4小时)滴加到溶液4中,维持体系温度在15℃,加毕,自然温度再持续搅拌2小时,过滤、打浆洗涤、烘干制得对二亚硝基苯产物,干重:65.99g,产率:80.74%(以对苯醌二肟为原料计算),灼烧残渣:0.74%。
[0096]
本实施例中第一步反应滴加时长为1小时,也可以是5小时或是1-5小时之间的任意时长;
[0097]
本实施例中在第三步反应合成前,对naclo溶液的有效氯进行测定,有效氯数值两天内有效;
[0098]
本实施例中的第一步和第三步的反应中的naoh也可以是koh、nahco3、na2co3、khco3、k2co3中的任何一种替代。
[0099]
实施例9:
[0100]
第一步,将亚硝酸钠(2.0mol,138.30g)和氢氧化钠(1.5mol,60.08g)加入483ml自来水,搅拌使之完全溶解,再加入苯酚(1.0mol,94.35g),搅拌溶解制得混合溶液1;将浓硫酸(3.0mol,294.09g)缓慢加入535ml冰水中,搅拌稀释制得溶液2;
[0101]
在3℃的环境温度下,将溶液1缓慢(5小时)滴加到溶液2中,维持体系温度在5℃,加毕再持续搅拌2小时,过滤、洗涤制得中间体对亚硝基苯酚湿料;
[0102]
第二步,将盐酸羟胺(2.0mol,139.16g)加入109ml自来水,搅拌溶解制得溶液3;在42℃的环境温度下,将溶液3(2小时)滴加到中间体对亚硝基苯酚湿料的水溶液中,维持体系温度在40℃,加毕再持续搅拌2小时,过滤、洗涤制得对苯醌二肟湿料(湿重:130.67g)。取样(5.2143g)置于鼓风烘箱,烘干至恒重(3.4279g),测得样品固含量:65.74%,对苯醌二肟产率:62.19%;
[0103]
第三步,将对苯醌二肟湿料(0.597mol,125.46g,湿料)溶解于氢氧化钠(1.09mol,43.44g)的水溶液(988ml)中,配置成碱性对苯醌二肟溶液4;将naclo溶液(有效氯7.10%,0.657mol,656.29g)加入276ml自来水,配置成有效氯为5%naclo溶液稀溶液5;在13℃的环境温度及反应容器完全避光处理的条件下,将溶液5(4小时)滴加到溶液4中,维持体系温度在15℃,加毕,自然温度再持续搅拌2小时,过滤、打浆洗涤、烘干制得对二亚硝基苯产物,干
重:66.16g,产率:81.41%(以对苯醌二肟为原料计算),灼烧残渣:0.64%。
[0104]
本实施例中第一步反应滴加时长为5小时,也可以是5小时或是1-5小时之间的任意时长;
[0105]
本实施例中在第三步反应合成前,对naclo溶液的有效氯进行测定,有效氯数值两天内有效;
[0106]
本实施例中的第一步和第三步的反应中的naoh也可以是koh、nahco3、na2co3、khco3、k2co3中的任何一种替代。
[0107]
实施例10:
[0108]
第一步,将亚硝酸钠(2.0mol,137.98g)和氢氧化钠(1.5mol,60.30g)加入483ml自来水,搅拌使之完全溶解,再加入苯酚(1.0mol,94.35g),搅拌溶解制得混合溶液1;将浓硫酸(3.0mol,294.31g)缓慢加入535ml冰水中,搅拌稀释制得溶液2;
[0109]
在3℃的环境温度下,将溶液1缓慢(2小时)滴加到溶液2中,维持体系温度在5℃,加毕再持续搅拌2小时,过滤、洗涤制得中间体对亚硝基苯酚湿料;
[0110]
第二步,将盐酸羟胺(2.0mol,139.00g)加入109ml自来水,搅拌溶解制得溶液3;在56℃的环境温度下,将溶液3(2小时)滴加到中间体对亚硝基苯酚湿料的水溶液中,维持体系温度在55℃,加毕再持续搅拌2小时,过滤、洗涤制得对苯醌二肟湿料(湿重:128.70g)。取样(5.1452g)置于鼓风烘箱,烘干至恒重(3.5198g),测得样品固含量:68.41%,对苯醌二肟产率:63.74%;
[0111]
第三步,将对苯醌二肟湿料(0.612mol,123.56g,湿料)溶解于氢氧化钠(1.11mol,44.52g)的水溶液(1017ml)中,配置成碱性对苯醌二肟溶液4;将naclo溶液(有效氯7.10%,0.673mol,672.59g)加入283ml自来水,配置成有效氯为5%naclo溶液稀溶液5;在13℃的环境温度及反应容器完全避光处理的条件下,将溶液5(4小时)滴加到溶液4中,维持体系温度在15℃,加毕,自然温度再持续搅拌2小时,过滤、打浆洗涤、烘干制得对二亚硝基苯产物,干重:68.50g,产率:82.24%(以对苯醌二肟为原料计算),灼烧残渣:0.78%。
[0112]
本实施例中第二步反应温度为55℃,也可以是70℃或是40℃-70℃之间任意温度;
[0113]
本实施例中在第三步反应合成前,对naclo溶液的有效氯进行测定,有效氯数值两天内有效;
[0114]
本实施例中的第一步和第三步的反应中的naoh也可以是koh、nahco3、na2co3、khco3、k2co3中的任何一种替代。
[0115]
实施例11:
[0116]
第一步,将亚硝酸钠(2.0mol,138.14g)和氢氧化钠(1.5mol,60.17g)加入483ml自来水,搅拌使之完全溶解,再加入苯酚(1.0mol,94.41g),搅拌溶解制得混合溶液1;将浓硫酸(3.0mol,294.1g)缓慢加入535ml冰水中,搅拌稀释制得溶液2;
[0117]
在5℃的环境温度下,将溶液1缓慢(2小时)滴加到溶液2中,维持体系温度在5℃,加毕再持续搅拌2小时,过滤、洗涤制得中间体对亚硝基苯酚湿料;
[0118]
第二步,将盐酸羟胺(2.0mol,139.04g)加入109ml自来水,搅拌溶解制得溶液3;在71℃的环境温度下,将溶液3(2小时)滴加到中间体对亚硝基苯酚湿料的水溶液中,维持体系温度在70℃,加毕再持续搅拌2小时,过滤、洗涤制得对苯醌二肟湿料(湿重:110.73g)。取
样(5.0241g)置于鼓风烘箱,烘干至恒重(3.8746g),测得样品固含量:77.12%,对苯醌二肟产率:61.82%;
[0119]
第三步,将对苯醌二肟湿料(0.5mol,105.70g,湿料)溶解于氢氧化钠(1.11mol,42.93g)的水溶液(1018.97ml)中,配置成碱性对苯醌二肟溶液4;将naclo溶液(有效氯6.87%,0.673mol,670.38g)加入250.72ml自来水,配置成有效氯为5%naclo溶液稀溶液5;在13℃的环境温度及反应容器完全避光处理的条件下,将溶液5(4小时)滴加到溶液4中,维持体系温度在15℃,加毕,自然温度再持续搅拌2小时,过滤、打浆洗涤、烘干制得对二亚硝基苯产物,干重:64.16g,产率:79.87%(以对苯醌二肟为原料计算),灼烧残渣:0.71%。
[0120]
本实施例中第二步反应温度为70℃,也可以是40℃-70℃之间任意温度;
[0121]
本实施例中在第三步反应合成前,对naclo溶液的有效氯进行测定,有效氯数值两天内有效;
[0122]
本实施例中的第一步和第三步的反应中的naoh也可以是koh、nahco3、na2co3、khco3、k2co3中的任何一种替代。
[0123]
实施例12:
[0124]
第一步,将亚硝酸钠(2.0mol,137.99g)和氢氧化钠(1.5mol,60.07g)加入483ml自来水,搅拌使之完全溶解,再加入苯酚(1.0mol,94.15g),搅拌溶解制得混合溶液1;将浓硫酸(3.0mol,294.31g)缓慢加入535ml冰水中,搅拌稀释制得溶液2;
[0125]
在3℃的环境温度下,将溶液1缓慢(2小时)滴加到溶液2中,维持体系温度在5℃,加毕再持续搅拌2小时,过滤、洗涤制得中间体对亚硝基苯酚湿料;
[0126]
第二步,将盐酸羟胺(2.0mol,139.12g)加入109ml自来水,搅拌溶解制得溶液3;在42℃的环境温度下,将溶液3(0.5小时)滴加到中间体对亚硝基苯酚湿料的水溶液中,维持体系温度在40℃,加毕再持续搅拌2小时,过滤、洗涤制得对苯醌二肟湿料(湿重:117.44g)。取样(5.2310g)置于鼓风烘箱,烘干至恒重(3.8767g),测得样品固含量:74.11%,对苯醌二肟产率:63.01%;
[0127]
第三步,将对苯醌二肟湿料(0.602mol,112.21g,湿料)溶解于氢氧化钠(1.09mol,43.80g)的水溶液(1010ml)中,配置成碱性对苯醌二肟溶液4;将naclo溶液(有效氯6.87%,0.662mol,683.89g)加入256ml自来水,配置成有效氯为5%naclo溶液稀溶液5;在13℃的环境温度及反应容器完全避光处理的条件下,将溶液5(4小时)滴加到溶液4中,维持体系温度在15℃,加毕,自然温度再持续搅拌2小时,过滤、打浆洗涤、烘干制得对二亚硝基苯产物,干重:65.34g,产率:79.74%(以对苯醌二肟为原料计算),灼烧残渣:0.62%,灼烧残渣:0.80%。
[0128]
本实施例中第二步反应滴加时长为0.5小时,也可以是0.5-2小时之间的任意时长;
[0129]
本实施例中在第三步反应合成前,对naclo溶液的有效氯进行测定,有效氯数值两天内有效;
[0130]
本实施例中的第一步和第三步的反应中的naoh也可以是koh、nahco3、na2co3、khco3、k2co3中的任何一种替代。
[0131]
实施例13:
[0132]
第一步,将亚硝酸钠(2.0mol,138.13g)和氢氧化钠(1.5mol,60.14g)加入483ml自
来水,搅拌使之完全溶解,再加入苯酚(1.0mol,94.15g),搅拌溶解制得混合溶液1;将浓硫酸(3.0mol,294.12g)缓慢加入535ml冰水中,搅拌稀释制得溶液2;
[0133]
在3℃的环境温度下,将溶液1缓慢(2小时)滴加到溶液2中,维持体系温度在5℃,加毕再持续搅拌2小时,过滤、洗涤制得中间体对亚硝基苯酚湿料;
[0134]
第二步,将盐酸羟胺(2.0mol,138.97g)加入109ml自来水,搅拌溶解制得溶液3;在42℃的环境温度下,将溶液3(2小时)滴加到中间体对亚硝基苯酚湿料的水溶液中,维持体系温度在40℃,加毕再持续搅拌2小时,过滤、洗涤制得对苯醌二肟湿料(湿重:124.03g)。取样(5.6871g)置于鼓风烘箱,烘干至恒重(4.0606g),测得样品固含量:71.40%,对苯醌二肟产率:64.11%;
[0135]
第三步,将对苯醌二肟湿料(0.612mol,118.34g,湿料)溶解于氢氧化钠(1.11mol,44.50g)的水溶液(1022ml)中,配置成碱性对苯醌二肟溶液4;将naclo溶液(有效氯6.51%,0.918mol,533.31g)加入161ml自来水,配置成有效氯为5%naclo溶液稀溶液5;在13℃的环境温度及反应容器完全避光处理的条件下,将溶液5(4小时)滴加到溶液4中,维持体系温度在15℃,加毕,自然温度再持续搅拌2小时,过滤、打浆洗涤、烘干制得对二亚硝基苯产物,干重:60.15g,产率:72.25%(以对苯醌二肟为原料计算),灼烧残渣:0.62%。
[0136]
本实施例中第三步底物次氯酸钠的配比为1.5个当量,也可以是0.8个当量或是0.8-1.5个当量之间的任意当量;
[0137]
本实施例中在第三步反应合成前,对naclo溶液的有效氯进行测定,有效氯数值两天内有效;
[0138]
本实施例中的第一步和第三步的反应中的naoh也可以是koh、nahco3、na2co3、khco3、k2co3中的任何一种替代。
[0139]
实施例14:
[0140]
第一步,将亚硝酸钠(2.0mol,138.11g)和氢氧化钠(1.5mol,60.10g)加入483ml自来水,搅拌使之完全溶解,再加入苯酚(1.0mol,94.31g),搅拌溶解制得混合溶液1;将浓硫酸(3.0mol,294.31g)缓慢加入535ml冰水中,搅拌稀释制得溶液2;
[0141]
在3℃的环境温度下,将溶液1缓慢(2小时)滴加到溶液2中,维持体系温度在5℃,加毕再持续搅拌2小时,过滤、洗涤制得中间体对亚硝基苯酚湿料;
[0142]
第二步,将盐酸羟胺(2.0mol,139.21g)加入109ml自来水,搅拌溶解制得溶液3;在42℃的环境温度下,将溶液3(2小时)滴加到中间体对亚硝基苯酚湿料的水溶液中,维持体系温度在40℃,加毕再持续搅拌2小时,过滤、洗涤制得对苯醌二肟湿料(湿重:123.15g)。取样(5.1413g)置于鼓风烘箱,烘干至恒重(3.6041g),测得样品固含量:70.10%,对苯醌二肟产率:62.50%;
[0143]
第三步,将对苯醌二肟湿料(0.599mol,118.01g,湿料)溶解于氢氧化钠(1.11mol,43.57g)的水溶液(999ml)中,配置成碱性对苯醌二肟溶液4;将naclo溶液(有效氯6.74%,0.479mol,945.62g)加入329ml自来水,配置成有效氯为5%naclo溶液稀溶液5;在13℃的环境温度及反应容器完全避光处理的条件下,将溶液5(4小时)滴加到溶液4中,维持体系温度在15℃,加毕,自然温度再持续搅拌2小时,过滤、打浆洗涤、烘干制得对二亚硝基苯产物,干重:62.46g,产率:76.62%(以对苯醌二肟为原料计算),灼烧残渣:0.54%。
[0144]
本实施例中第三步底物次氯酸钠的配比为0.8个当量,也可以是0.8-1.5个当量之
间的任意当量;
[0145]
本实施例中在第三步反应合成前,对naclo溶液的有效氯进行测定,有效氯数值两天内有效;
[0146]
本实施例中的第一步和第三步的反应中的naoh也可以是koh、nahco3、na2co3、khco3、k2co3中的任何一种替代。
[0147]
实施例15:
[0148]
第一步,将亚硝酸钠(2.0mol,138.05g)和氢氧化钠(1.5mol,60.07g)加入483ml自来水,搅拌使之完全溶解,再加入苯酚(1.0mol,94.09g),搅拌溶解制得混合溶液1;将浓硫酸(3.0mol,294.21g)缓慢加入535ml冰水中,搅拌稀释制得溶液2;
[0149]
在3℃的环境温度下,将溶液1缓慢(2小时)滴加到溶液2中,维持体系温度在5℃,加毕再持续搅拌2小时,过滤、洗涤制得中间体对亚硝基苯酚湿料;
[0150]
第二步,将盐酸羟胺(2.0mol,139.28g)加入109ml自来水,搅拌溶解制得溶液3;在42℃的环境温度下,将溶液3(2小时)滴加到中间体对亚硝基苯酚湿料的水溶液中,维持体系温度在40℃,加毕再持续搅拌2小时,过滤、洗涤制得对苯醌二肟湿料(湿重:144.13g)。取样(5.0403g)置于鼓风烘箱,烘干至恒重(3.0161g),测得样品固含量:59.84%,对苯醌二肟产率:62.44%;
[0151]
第三步,将对苯醌二肟湿料(0.603mol,139.09g,湿料)溶解于氢氧化钠(1.10mol,43.84g)的水溶液(985ml)中,配置成碱性对苯醌二肟溶液4;将naclo溶液(有效氯6.21%,0.663mol,757.23g)加入183ml自来水,配置成有效氯为5%naclo溶液稀溶液5;在26℃的环境温度及反应容器完全避光处理的条件下,将溶液5(4小时)滴加到溶液4中,维持体系温度在25℃,加毕,自然温度再持续搅拌2小时,过滤、打浆洗涤、烘干制得对二亚硝基苯产物,干重:64.83g,产率:79.05%(以对苯醌二肟为原料计算),灼烧残渣:0.67%。
[0152]
本实施例中第二步反应温度为25℃,也可以是5℃或是5℃-25℃之间任意温度;
[0153]
本实施例中在第三步反应合成前,对naclo溶液的有效氯进行测定,有效氯数值两天内有效;
[0154]
本实施例中的第一步和第三步的反应中的naoh也可以是koh、nahco3、na2co3、khco3、k2co3中的任何一种替代。
[0155]
实施例16:
[0156]
第一步,将亚硝酸钠(2.0mol,138.01g)和氢氧化钠(1.5mol,60.03g)加入483ml自来水,搅拌使之完全溶解,再加入苯酚(1.0mol,94.09g),搅拌溶解制得混合溶液1;将浓硫酸(3.0mol,294.41g)缓慢加入535ml冰水中,搅拌稀释制得溶液2;
[0157]
在3℃的环境温度下,将溶液1缓慢(2小时)滴加到溶液2中,维持体系温度在5℃,加毕再持续搅拌2小时,过滤、洗涤制得中间体对亚硝基苯酚湿料;
[0158]
第二步,将盐酸羟胺(2.0mol,139.13g)加入109ml自来水,搅拌溶解制得溶液3;在42℃的环境温度下,将溶液3(2小时)滴加到中间体对亚硝基苯酚湿料的水溶液中,维持体系温度在40℃,加毕再持续搅拌2小时,过滤、洗涤制得对苯醌二肟湿料(湿重:119.44g)。取样(5.4102g)置于鼓风烘箱,烘干至恒重(3.9505g),测得样品固含量:73.02%,对苯醌二肟产率:63.14%;
[0159]
第三步,将对苯醌二肟湿料(0.603mol,114.03g,湿料)溶解于氢氧化钠(1.10mol,
43.85g)的水溶液(1010ml)中,配置成碱性对苯醌二肟溶液4;将naclo溶液(有效氯6.51%,0.663mol,940.85g)加入218.23ml自来水,配置成有效氯为5%naclo溶液稀溶液5;在4℃的环境温度及反应容器完全避光处理的条件下,将溶液5(4小时)滴加到溶液4中,维持体系温度在5℃,加毕,自然温度再持续搅拌2小时,过滤、打浆洗涤、烘干制得对二亚硝基苯产物,干重:61.13g,产率:74.50%(以对苯醌二肟为原料计算),灼烧残渣:0.36%。
[0160]
本实施例中第二步反应温度为5℃,也可以是5℃-25℃之间任意温度;
[0161]
本实施例中在第三步反应合成前,对naclo溶液的有效氯进行测定,有效氯数值两天内有效;
[0162]
本实施例中的第一步和第三步的反应中的naoh也可以是koh、nahco3、na2co3、khco3、k2co3中的任何一种替代。
[0163]
实施例17:
[0164]
第一步,将亚硝酸钠(2.0mol,138.12g)和氢氧化钠(1.5mol,60.13g)加入483ml自来水,搅拌使之完全溶解,再加入苯酚(1.0mol,94.27g),搅拌溶解制得混合溶液1;将浓硫酸(3.0mol,294.14g)缓慢加入535ml冰水中,搅拌稀释制得溶液2;
[0165]
在3℃的环境温度下,将溶液1缓慢(2小时)滴加到溶液2中,维持体系温度在5℃,加毕再持续搅拌2小时,过滤、洗涤制得中间体对亚硝基苯酚湿料;
[0166]
第二步,将盐酸羟胺(2.0mol,139.01g)加入109ml自来水,搅拌溶解制得溶液3;在42℃的环境温度下,将溶液3(2小时)滴加到中间体对亚硝基苯酚湿料的水溶液中,维持体系温度在40℃,加毕再持续搅拌2小时,过滤、洗涤制得对苯醌二肟湿料(湿重:125.88g)。取样(5.0014g)置于鼓风烘箱,烘干至恒重(3.4690g),测得样品固含量:69.36%,对苯醌二肟产率:63.21%;
[0167]
第三步,将对苯醌二肟湿料(0.607mol,120.88g,湿料)溶解于氢氧化钠(1.10mol,44.16g)的水溶液(1011ml)中,配置成碱性对苯醌二肟溶液4;将naclo溶液(有效氯6.01%,0.668mol,788.18g)加入159ml自来水,配置成有效氯为5%naclo溶液稀溶液5;在13℃的环境温度及反应容器完全避光处理的条件下,将溶液5(1小时)滴加到溶液4中,维持体系温度在15℃,加毕,自然温度再持续搅拌2小时,过滤、打浆洗涤、烘干制得对二亚硝基苯产物,干重:66.43g,产率:80.41%(以对苯醌二肟为原料计算),灼烧残渣:0.76%。
[0168]
本实施例中第三步反应滴加时长为1小时,也可以是1-4小时之间的任意时长;
[0169]
本实施例中在第三步反应合成前,对naclo溶液的有效氯进行测定,有效氯数值两天内有效;
[0170]
本实施例中的第一步和第三步的反应中的naoh也可以是koh、nahco3、na2co3、khco3、k2co3中的任何一种替代。
[0171]
对比例1:
[0172]
将对苯醌二肟(1.086mol,150.14g,干料)溶解于氢氧化钠(1.97mol,78.91g)的水溶液(1875ml)中,配置成碱性对苯醌二肟溶液4;将naclo溶液(有效氯6.57%,1.195mol,1694.96g)加入405ml自来水,配置成有效氯为5%naclo溶液稀溶液5;在13℃的环境温度及反应容器完全避光处理的条件下,将溶液5(4小时)滴加到溶液4中,维持体系温度在15℃,加毕,自然温度再持续搅拌2小时,过滤、打浆洗涤、烘干制得对二亚硝基苯产物,干重:118.91g,产率:80.45%(以对苯醌二肟为原料计算),灼烧残渣:0.69%。
[0173]
本对比例中使用中间体对苯醌二肟为采购粉末产品;
[0174]
本对比例中在反应合成前,对naclo溶液的有效氯进行测定,有效氯数值两天内有效;
[0175]
本对比例反应中的naoh也可以是koh、nahco3、na2co3、khco3、k2co3中的任何一种替代。
[0176]
实施例1-实施例17及对比例1所得对二亚硝基苯样品的收率、外观性状及所测试的理化指标数据见表1;实施例1-实施例17及对比例1所得对二亚硝基苯样品的应用性能评估数据见表2;实施例1-实施例8所得的对二亚硝基苯样品的红外谱图数据见图1和图2;实施例9-实施例17所得的对二亚硝基苯样品的红外谱图数据见图3和图4;实施例1和对比例1所得的对二亚硝基苯样品的红外谱图数据见图5和图6。
[0177]
表1实施例1-实施例17以及对比例1对二亚硝基苯样品的数据及理化指标测试
[0178][0179]
从表1可见,实施例1至实施例12的实施例中,在未改变第三步的反应条件的情况下,实施例4减少硫酸的配比当量,会导致最终产物对二亚硝基苯产率的降低;其他条件的改变,对最终产物对二亚硝基苯的产率及外观性状的影响并不大。对比于实施例1,苯酚与其他底物配比当量的减少会降低中间体对苯醌二肟的产率,尤其是亚硝酸钠、硫酸当量的降低;第一步亚硝化过程温度的降低和滴加速度的改变,第二步缩合温度的提高和滴加速度的提升对产率中间体对苯醌二肟的产率影响不大,但产率均不如实施例1。
[0180]
相较与对比例1,在第三步反应条件完全相同的实施例中,除了实施例4样品的收率、外观不同外,其他实施例对苯醌二肟湿料与干料投料所得产物样品的产率、外观、灼烧残渣均没有明显区别。
[0181]
实施例13至实施例17的实施例中,在未改变前两步的反应条件的情况下,次氯酸钠的配比和反应的温度会影响产率的变动及产物的外观性状,尤其是配比的增大及温度的提高会使产物颜色明显变深。
[0182]
所有实施例或对比例产物样品的灼烧残渣均≤0.8%,数据比较可观。
[0183]
表2实施例1-实施例17以及对比例1对二亚硝基苯样品的应用性能评估数据
[0184][0185]
注:1.将所有实施例的样品与对比例1样品制样评测其粘接性能;
[0186]
2.r为粘接剥离后粘接面上橡胶附胶比例,理想的破环形式是100r。
[0187]
从表2可见,所有实施例以及对比例的样品粘接测试性能均表现优异;实施例1至实施例12的实施例中,在未改变第三步的反应条件及相同的性能评估测试条件的情况下,实施例所测得对二亚硝基苯样品粘接性能的数据,均与对比例1样品数据没有明显区别;实施例13至实施例17的实施例中,在改变第三步的反应条件的情况下,所测得对二亚硝基苯样品粘接性能的数据,实施例13、实施例15的样品以三元乙丙橡胶作为粘合剂测得的性能数据相对较差。
[0188]
从图1、图2、图3、图4可见,实施例1至实施例12的实施例中,在未改变第三步的反应条件的情况下,观察实施例样品的红外谱图数据峰型及特殊吸收峰区域(硝基官能团n=o的对称、反对称伸缩振动吸收峰),前两步反应条件的改变对最终产物对二亚硝基苯红外数据影响并不大;实施例13至实施例17的实施例中,改变第三步的反应条件,样品对二亚硝基苯的红外谱图变化非常明显,次氯酸钠配比当量的过量、温度的提升或反滴加时间过快均会导致最终产物的过度氧化,使致红外谱图数据中硝基的特殊吸收峰区域的凸显以及红外谱图的重叠性变得较差。
[0189]
从图5、图6可见对比例1与实施例1对比,在相同的反应条件下,所得到的样品红外谱图几乎完全重叠。
[0190]
本领域技术人员在考虑说明书及实践这里公开的公开后,将容易想到本技术的其它实施方案。本技术旨在涵盖本技术的任何变型、用途或者适应性变化,这些变型、用途或者适应性变化遵循本技术的一般性原理并包括本技术未公开的本技术领域中的公知常识或惯用技术手段。说明书和实施例仅被视为示例性的,本技术的真正范围和精神由下面的权利要求指出。
[0191]
应当理解的是,本技术并不局限于上面已经描述的精确结构,并且可以在不脱离其范围进行各种修改和改变。本技术的范围仅由所附的权利要求来限制。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1