一种异紫蒽酮的制备方法与流程
:
1.本发明专利涉及有机还原染料技术领域,具体涉及一种异紫蒽酮的制备方法。
背景技术:
2.异紫蒽酮,俗称还原紫10,溶于邻氯苯酚、二甲苯为红色,有荧光,微溶于丙酮、氯仿、吡啶和甲苯,不溶于乙醇。在浓硫酸中为暗绿色,稀释后有紫色沉淀;在碱性保险粉溶液中为暗红光蓝色;在酸性保险粉溶液中为暗红色。主要用于棉及其混纺织物的染色、直接印花、防染印花、拔染印花,也可用于粘胶纤维的染色。现有的制备异紫蒽酮的方法是将苯绕蒽酮溴化后得到3-溴苯绕蒽酮,3-溴苯绕蒽酮转化为相应的硫化物(联硫苯绕蒽酮),联硫苯绕蒽酮用碱/正丁醇混合溶液处理,离析,氧化得到异紫蒽酮。上述制备异紫蒽酮的过程中会产生大量的含硫废水,对环境很不友好,极易对环境造成污染,同时过量的碱被浪费,增加了生产成本。
技术实现要素:
3.本发明的目的在于提供一种反应过程简单,实现零排放的异紫蒽酮的制备方法。
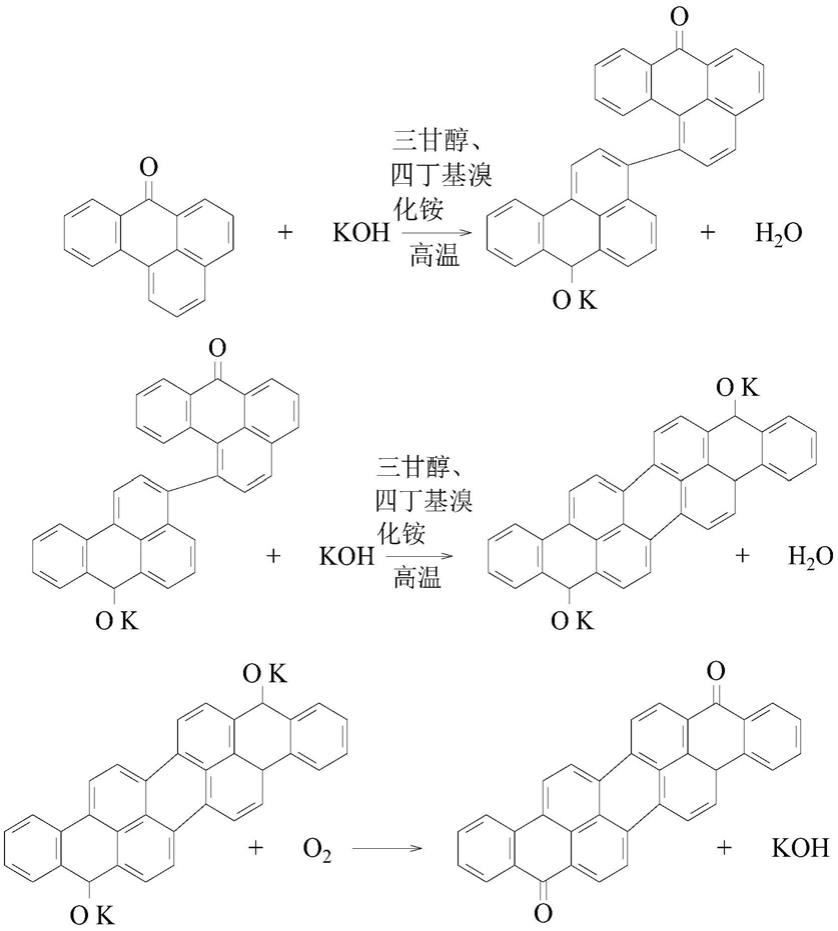
4.本发明的目的由如下技术方案实施:一种异紫蒽酮的制备方法,其依次包括有如下步骤:
5.(1)制备混合液:向反应瓶中加入氢氧化钾、三甘醇和四丁基溴化铵混合均匀,接着升温至150-220℃,保温0.5-1h,目的使混合更充分,得到混合液;
6.(2)氧化还原反应:向步骤(1)中的所述混合液中加入苯绕蒽酮,并通入压缩空气,然后继续保温2-6h,保温完成后,停止通入所述压缩空气,并自然冷却至50-100℃,得到反应物料;
7.以三甘醇为助溶剂,四丁基溴化铵为定位剂和催化剂,具体反应方程式如下:
[0008][0009]
(3)制备异紫蒽酮:将步骤(2)中的所述反应物料置于质量是所述反应物料8-11倍的水中进行搅拌稀释,然后经过过滤得到母液和固体物料,所述固体物料再经过洗涤后烘干,得到异紫蒽酮,洗涤后的水回用作清洗所述反应物料。
[0010]
进一步的,所述苯绕蒽酮与所述氢氧化钾的质量比为1:2-10。
[0011]
进一步的,所述苯绕蒽酮与所述三甘醇的质量比为1:1-10。
[0012]
进一步的,添加的所述四丁基溴化铵为所述苯绕蒽酮重量的1%-5%。
[0013]
进一步的,所述步骤(1)中优选温度170-190℃。
[0014]
进一步的,在所述步骤(2)中,将所述苯绕蒽酮至少均匀分为12次加入所述混合液中,每次当所述混合液温度降至170℃时添加所述苯绕蒽酮,避免一次添加过多导致溶液粘稠,进而无法充分反应。
[0015]
进一步的,所述步骤(2)中的所述压缩空气的压力为0.025-0.05mpa。
[0016]
进一步的,所述步骤(3)中的搅拌时间为1.5-5h,搅拌转速为500-1000rpm。
[0017]
进一步的,所述步骤(3)中所述母液的处理方法,具体包括以下步骤:
[0018]
a、蒸发浓缩:将所述母液进行蒸发浓缩,直至溶液的碱度达到40-50%时,停止蒸
发浓缩进行过滤,得到固体物和原料溶液;
[0019]
b、干燥:将所述原料溶液进行干燥,干燥温度为190-210℃,直至水分含量低于1%,停止干燥,得到氢氧化钾、三甘醇和四丁基溴化铵的混合原料,可以套用于下一次反应。
[0020]
进一步的,所述步骤(3)中的烘干温度为105-125℃。
[0021]
本发明的优点:
[0022]
1、本发明提供了一种异紫蒽酮的制备方法,以三甘醇为助溶剂,四丁基溴化铵为定位剂和催化剂,苯绕蒽酮与氢氧化钾反应得到异紫蒽酮隐色体中间产物和水,然后异紫蒽酮隐色体中间产物在与氧气反应得到异紫蒽酮和氢氧化钾,反应过程简单,提高了生产效率,并且中间体废水的产量减少,进而减少了废水的处理量。
[0023]
2、本发明提供了一种异紫蒽酮的制备方法,经过反应后所产生的废弃污染物单一,只有少量的碳酸钾和少量的有机物杂质通过蒸发浓缩后析出,接着将其直接进行危废处理,处理量少;剩余的原料溶液经过干燥后得到氢氧化钾、三甘醇和四丁基溴化铵的混合原料,可以套用于下一次反应,基本实现了零排放,实现了绿色生产的目标,同时避免了原料浪费,降低了生产成本。
具体实施方式:
[0024]
下面将通过实施例对本发明作进一步的详细说明。
[0025]
实施例1:向反应瓶中加入120g氢氧化钾、80g三甘醇和0.6g四丁基溴化铵混合均匀,接着升温至170-190℃,保温1h,目的使混合更充,得到混合液;然后将60g苯绕蒽酮均匀分为24次,每次当混合液温度降至170℃时添加苯绕蒽酮,避免一次添加过多导致溶液粘稠,进而无法充分反应,在添加苯绕蒽酮的过程中一直通入压力为0.025mpa的压缩空气,然后继续保温3h,保温完成后,停止通入压缩空气,并自然冷却至50-100℃,得到133ml反应物料,将反应物料置于700ml的水中进行3h的搅拌稀释,搅拌速度为700rpm,然后经过过滤得到0.8l母液和固体物料;固体物料再经过洗涤后在105-125℃下烘干,得到59.5g异紫蒽酮;前期200ml的洗涤水与母液一起浓缩蒸发,后期700ml的洗涤水用作稀释反应物料。
[0026]
对1l的母液及前期洗涤水混合液进行蒸发浓缩,直至溶液的碱度达到40-50%时,停止蒸发浓缩进行过滤,得到6g固体物和0.24l原料溶液;6g的固体物作为危废被处理,原料溶液则被干燥,干燥温度为190-210℃,直至水分含量低于1%,停止干燥,干燥后得到氢氧化钾、三甘醇和四丁基溴化铵的混合原料,可以套用于下一次反应。
[0027]
实施例2:向反应瓶中加入600g氢氧化钾、600g三甘醇和3g四丁基溴化铵混合均匀,接着升温至170-190℃,保温1h,目的使混合更充,得到混合液;然后将60g苯绕蒽酮均匀分为12次,每次当混合液温度降至170℃时添加苯绕蒽酮,避免一次添加过多导致溶液粘稠,进而无法充分反应,在添加苯绕蒽酮的过程中一直通入压力为0.025mpa的压缩空气,然后继续保温3h,保温完成后,停止通入压缩空气,并自然冷却至50-100℃,得到800ml反应物料,将反应物料置于3000ml的水中进行5h的搅拌稀释,搅拌速度为500rpm,然后经过过滤得到3.4l母液和固体物料;固体物料再经过洗涤后在105-125℃下烘干,得到58.7g异紫蒽酮;前期200ml的洗涤水与母液一起浓缩蒸发,后期800ml的洗涤水用作稀释反应物料。
[0028]
对3.6l的母液及前期洗涤水混合液进行蒸发浓缩,直至溶液的碱度达到40-50%
时,停止蒸发浓缩进行过滤,得到27g固体物和1.2l原料溶液;27g的固体物作为危废被处理,原料溶液则被干燥,干燥温度为190-210℃,直至水分含量低于1%,停止干燥,干燥后得到氢氧化钾、三甘醇和四丁基溴化铵的混合原料,可以套用于下一次反应。
[0029]
实施例3:向反应瓶中加入360g氢氧化钾、60g三甘醇和1.8g四丁基溴化铵混合均匀,接着升温至170-190℃,保温1h,目的使混合更充,得到混合液;然后将60g苯绕蒽酮均匀分为30次,每次当混合液温度降至170℃时添加苯绕蒽酮,避免一次添加过多导致溶液粘稠,进而无法充分反应,在添加苯绕蒽酮的过程中一直通入压力为0.025mpa的压缩空气,然后继续保温3h,保温完成后,停止通入压缩空气,并自然冷却至50-100℃,得到120ml反应物料,将反应物料置于2400ml的水中进行30min的搅拌稀释,搅拌速度为700rpm,然后经过过滤得到2.4l母液和固体物料;固体物料再经过洗涤后在105-125℃下烘干,得到59.1g异紫蒽酮;前期200ml的洗涤水与母液一起浓缩蒸发,后期900ml的洗涤水用作稀释反应物料。
[0030]
对2.6l的母液及前期洗涤水混合液进行蒸发浓缩,直至溶液的碱度达到40-50%时,停止蒸发浓缩进行过滤,得到12.3g固体物和0.72l原料溶液;12.3g的固体物作为危废被处理,原料溶液则被干燥,干燥温度为190-210℃,直至水分含量低于1%,停止干燥,干燥后得到氢氧化钾、三甘醇和四丁基溴化铵的混合原料,可以套用于下一次反应。
[0031]
对比例1:
[0032]
现有的制备异紫蒽酮的方法,具体包括以下步骤:
[0033]
(1)3-溴代苯绕蒽酮的合成
[0034]
向四口瓶中加入1500ml水,投入苯绕蒽酮150g,加入邻二氯苯35g,搅拌分散8h。,加入硫酸,控制酸度4.2-4.7%为合格,否则调之。加入86g溴素,用约60min均匀放入溴化锅,控制温度35℃以下,继续搅拌15min,在2h内均匀升温至71-73℃,保持15min。在2.5-3.5h的时间均匀滴加将250ml20%氯酸钠水溶液,并保温1h,得到3-溴代苯绕蒽酮溶液,经过过滤得到3-溴代苯绕蒽酮,以及废水。
[0035]
(2)硫代反应
[0036]
加入硫指数为1.85-2.10,溶度为8%的多硫化钠2500ml,投入折干品600g 3-溴代苯绕蒽酮,升温至90-100℃,打浆1h,逐步升温122-128℃并保持9h,降温至80-90℃,加入10%的氢氧化钠溶液300ml,压料、洗涤,母液及前期洗水进入母液罐,后期洗水套用梯度洗涤,物料烘干,得到联硫苯绕蒽酮,再次过程中产生的废水有10000g,固废4g。
[0037]
(3)缩合、氧化、压料
[0038]
在缩合釜中投入联硫苯绕蒽酮550g,加入2100ml丁醇,充分打浆分散均匀后,投入700g氢氧化钾,升温至112-125℃并保持4h。保温结束后,控制温度在120-140℃,边丁醇边加入水,液面无油珠,降温加水稀释至4500ml,通入压缩空气氧化直至渗圈无色。压滤洗涤,烘干最后得到512g异紫蒽酮,再次过程中产生的废水有13816g。
[0039]
上述四个反应中均产生了大量的废水,并且该工艺方法复杂,进而制备异紫蒽酮的成本高,而本发明的实施例1中公开的方法流程简单,并未产生废水,只是产生少量的固废,基本实现了零排放,实现了绿色生产的目标,同时降低了生产异紫蒽酮的成本。
[0040]
以上是本发明的优选实施方式,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1