一种Etelcalcetide的合成方法及其应用与流程
一种etelcalcetide的合成方法及其应用
技术领域
1.本发明属于药物合成技术领域,具体涉及一种etelcalcetide的合成方法及其应用。
背景技术:
2.继发性甲状旁腺功能亢进症(shpt)是由于钙磷代谢异常、维生素d缺乏或肾功能损害引起的甲状旁腺增生,血甲状旁腺激素升高,进而加重骨骼损害的疾病。目前治疗shpt的措施包括积极控制诱发shpt的原发病,限制饮食中磷的摄入,必要时才有磷结合剂、活性维生素d类似物。同时拟钙剂能有效治疗shpt。
3.依特卡肽(etelcalcetide)是一种用于治疗接受血液透析患者shpt的拟钙药。在每次透析期结束时静脉注射给药。其通过结合并激活甲状旁腺中的钙敏感受体(casr)作为变构激活剂起作用,抑制并减少甲状旁腺素的分泌,从而达到可以治疗shpt的目的。该药也是首个在患者每次透析结束后每周三次静脉注射给药的静脉注射型拟钙剂。中重度甲状旁腺功能亢进血液透析患者接受维拉卡肽治疗后,pth水平显著降低。etelcalcetide是一种仿钙剂,可对钙敏感受体(casr)进行变构调节。etelcalcetide主链由4个d型arg、2个d型ala以及乙酰化d型cys共7个d型氨基酸组成,同时侧链通过二硫键与l-半胱氨酸相连,得到etelcalcetide。etelcalcetide结构如图1所示,肽序如图2所示。
4.国内外对etelcalcetide的合成已有报道,如公开号为cn105504012 a、cn106928320、cn109280078b、cn110668984a、cn110498835a以及cn110054662a的专利均采用固相合成etelcalcetide。虽然固相偶联过程操作过程简单、便于自动化生产、产品收率较高以及产品易分离的优点,但是偶联过程中需要使用树脂,为保证偶联完全,氨基酸的投料倍数通常需要过量(3-5当量),加上etelcalcetide主链均为d型氨基酸,价格相对常规氨基酸来说,比较昂贵。因此,采用固相合成方法,成本过高,在生产上规模化受到限制。
5.公开号为cn106928321、wo2016154580a1、cn111925418a的专利均采用液相合成etelcalcetide。液相合成的方法在适合大规模生产、氨基酸用量较少,达到控制成本的目的。但是反应后处理操作繁杂、难溶性肽合成较困难、总收率相对较低,并且在液相中形成多肽链间二硫键,会出现二硫键错配问题,副反应和副产物的种类较多,收率和纯度都不理想,进一步减低产品收率。
技术实现要素:
6.针对上述现有技术中存在的问题,本发明的目的在于设计提供一种etelcalcetide的合成方法及其应用。本发明利用液相载体方法合成etelcalcetide合成以及使用两个二肽片段提高合成效率和减少缺损杂质。本发明结合了固相合成和液相合成的优点,达到了操作简单、减低成本、减少缺损杂质的产生以及适合规模化生产的目的。并且可操作性好,杂质减少的效果良好,同时也提高了收率,降低了成本,适合规模化生产。
7.为了实现上述目的,本发明采用以下技术方案:
8.一种etelcalcetide的合成方法,其特征在于包括以下步骤:
9.(1)取氨基酸置于溶剂中进行偶联和重结晶,液相合成fmoc-d-ala-d-arg(pbf)-oh二肽片段;
10.(2)取氨基酸置于溶剂中进行偶联和重结晶,液相合成fmoc-d-arg(pbf)-d-arg(pbf)-oh二肽片段;
11.(3)以dpm-nh2为液相合成载体,以氯仿为反应溶剂,依次偶联以下氨基酸:上述步骤(1)合成的fmoc-d-ala-d-arg(pbf)-oh、上述步骤(2)合成的fmoc-d-arg(pbf)-d-arg(pbf)-oh、上述步骤(1)合成的fmoc-d-ala-d-arg(pbf)-oh、n-ac-d-cys(mmt)-oh,获得n-ac-d-cys(mmt)-d-ala-d-arg(pbf)-d-arg(pbf)-d-arg(pbf)-d-ala-d-arg(pbf)-dpm-nh2,即片段a,结构式如下式1;
[0012][0013]
(4)采用1%tfa/chcl3溶液脱除片段a中cys(mmt)的保护基,获得n-ac-d-cys(sh)-d-ala-d-arg(pbf)-d-arg(pbf)-d-arg(pbf)-d-ala-d-arg(pbf)-dpm-nh2,即片段b,再与boc-cys(npys)-oh偶联后,裂解获得etelcalcetide粗品。
[0014]
所述的一种etelcalcetide的合成方法,所述步骤(1)和步骤(2)中溶剂包括nmp、thf、dcm、acn和dmf中的一种或多种,所述偶联的体系包括dic/honb/有机碱、dcc/honb/有机碱、edci/hosu/有机碱、dcc/hosu/有机碱、dic/honb/无机碱、dcc/honb/无机碱中的一种或多种,所述重结晶的溶剂体系包括etoh/h2o、dcm/et2o、thf/et2o、ea/et2o、meoh/et2o、ch3cn/et2o、ea/pe、thf/pe、ch3cn/h2o中的一种或多种。
[0015]
所述的一种etelcalcetide的合成方法,所述有机碱包括dipea、三乙胺、n-甲基吗啡啉中的至少一种,优选有机碱为dipea,所述无机碱包括na2co3或nahco3中的至少一种,优选无机碱为nahco3。
[0016]
所述的一种etelcalcetide的合成方法,所述步骤(1)中溶剂为dcm,所述偶联的体系为dic/honb/有机碱,所述重结晶的溶剂体系为etoh/h2o。
[0017]
所述的一种etelcalcetide的合成方法,所述步骤(2)中溶剂为dcm,所述偶联的体系为dic/honb/有机碱,所述重结晶的溶剂体系为ea/pe。
[0018]
所述的一种etelcalcetide的合成方法,所述步骤(3)中氨基酸与dpm-nh2的摩尔比1:1~3:1,优选氨基酸与dpm-nh2的摩尔比为1.1:1~1.3:1,所述偶联采用的偶联剂包括edci//hobt、dipcdi/hobt、pybop/hobt/dipea、hbtu/hobt/dipea、dipcdi/hoat、hatu/hoat/dipea和pyaop/hoat/dipea中的一种或多种,优选偶联采用的偶联剂为edci//hobt。
[0019]
所述的一种etelcalcetide的合成方法,所述步骤(4)中cys(mmt)的保护基中mmt的体积为tfa/chcl3溶液的体积的1%-5%,优选cys(mmt)的保护基中mmt的体积为tfa/chcl3溶液的体积的1%-3%。
[0020]
所述的一种etelcalcetide的合成方法,所述步骤(4)中裂解采用的裂解试剂包括tfa、phsme、tis、phoh、h2o、phome中的一种或几种的混合物,优选裂解采用的裂解试剂为
tfa:h2o:phsme:phome:tis的体积比为88:5:3:2:2。
[0021]
一种etelcalcetide,是采用如权利要求1-8任一所述的etelcalcetide的合成方法合成得到的。
[0022]
任一所述的一种etelcalcetide的合成方法在合成etelcalcetide药物上的应用。
[0023]
与现有技术相比,本发明具有以下有益效果:
[0024]
1、本发明使用液相载体dpm-nh2,方法为均相反应,可以应用于亲水性、疏水性多肽、长链多肽的直接修饰同时可以在有机溶剂中进行,克服了固相多肽合成规模受限制的缺点,适合大规模化生产,提高了生产效率。
[0025]
2、本发明每步反应所用氨基酸当量为1.1eqv,与固相多肽合成方法相比,提高了氨基酸的利用率,节约了成本。每步反应中间体都可以用极性有机溶剂进行沉淀,与传统的液相多肽合成方法相比,后处理操作简单,每步中间体的纯度相对较高。
[0026]
3、本发明使用fmoc-d-ala-d-arg-oh、fmoc-d-arg-d-arg-oh两个二肽片段作为起始物料,进一步提高偶联效率。大大减少了合成步骤的同时,减少缺损arg杂质的产生,降低该肽的合成难度。
附图说明
[0027]
图1为etelcalcetide的结构图;
[0028]
图2为etelcalcetide的肽序图;
[0029]
图3为dpm-nh2的结构式;
[0030]
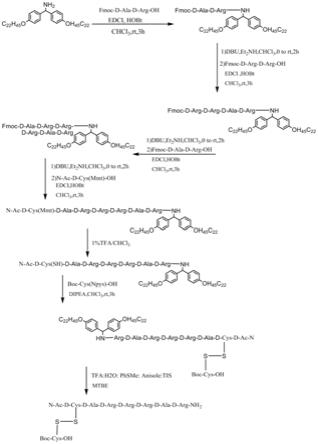
图4为etelcalcetide的合成路线图;
[0031]
图5为精肽的hplc图;
[0032]
图6为精肽的质谱图。
具体实施方式
[0033]
以下将通过附图和实施例对本发明作进一步说明。
[0034]
实施例1:fmoc-d-ala-d-arg(pbf)-oh的合成
[0035]
取200ml thf溶解fmoc-d-ala-oh(15.6g,50mmol)、hosu(6.33g,55mmol),冰水浴的条件下加入dcc(11.3g,55mmol),撤去冰浴,室温搅拌5h。tlc显示原料基本反应完全(pe:ea:hoac=1:1:0.05)反应混合物过滤,滤液备用。
[0036]
200ml去离子水溶解na2co3(15.9g,150mmol)、h-d-arg(pbf)-oh(21.32g,50mmol),缓慢加入上面新制备的混合液,室温搅拌18h。反应液浓缩,加入200ml去离子水稀释残留液,ea提取(250*3ml),水层1mol/l盐酸溶液酸化至ph约3,ea萃取,合并ea层,ea层依次用200ml 1mol/l盐酸溶液、200ml*2ml去离子水洗涤2次、200ml用饱和食盐水洗涤1次,用40g无水硫酸钠快速干燥,过滤浓缩,浓缩液用etoh/h2o(2:1)重结晶。产品过滤,真空干燥得到fmoc-d-ala-d-arg(pbf)-oh:24.9g,hplc:98.2%,收率:69.4%。
[0037]
实施例2:fmoc-d-ala-d-arg(pbf)-oh的合成
[0038]
取200ml dcm溶解fmoc-d-ala-oh(15.6g,50mmol)、honb(9.84g,55mmol),冰水浴的条件下加入dic(8.5ml,55mmol),撤去冰浴,室温搅拌5h。tlc显示原料基本反应完全(pe:ea:hoac=1:1:0.05)反应混合物过滤,滤液备用。
[0039]
将h-d-arg(pbf)-oh(21.32g,50mmol)加入上述滤液中,开始磁力搅拌,并用滴液漏斗滴加dipea(10.74ml,65mmol),室温搅拌10h。反应液浓缩,加入200ml去离子水稀释残留液,ea提取(250*3ml),水层1mol/l盐酸溶液酸化至ph约3,ea萃取,合并ea层,ea层依次用200ml 1mol/l盐酸溶液、200ml*2ml去离子水洗涤2次、200ml用饱和食盐水洗涤1次,用40g无水硫酸钠快速干燥,过滤浓缩,浓缩液用etoh/h2o(1:4)重结晶。产品过滤,真空干燥得到fmoc-d-ala-d-arg(pbf)-oh:26.7g,hplc:98.9%,收率:73.8%。
[0040]
实施例3:fmoc-d-arg(pbf)-d-arg(pbf)-oh的合成
[0041]
取200ml thf溶解fmoc-d-arg(pbf)-oh(32.4g,50mmol)、hosu(6.33g,55mmol),冰水浴的条件下加入dcc(11.3g,55mmol),撤去冰浴,室温搅拌5h。tlc显示原料基本反应完全(pe:ea:hoac=1:1:0.05)反应混合物过滤,滤液备用。
[0042]
200ml去离子水溶解na2co3(15.9g,150mmol)、h-d-arg(pbf)-oh(21.32g,50mmol),缓慢加入上面新制备的混合液,室温搅拌18h。反应液浓缩,加入200ml去离子水稀释残留液,ea提取(250*3ml),水层1mol/l盐酸溶液酸化至ph约3,ea萃取,合并ea层,ea层依次用200ml 1mol/l盐酸溶液、200ml*2ml去离子水洗涤2次、200ml用饱和食盐水洗涤1次,用40g无水硫酸钠快速干燥,过滤浓缩,浓缩液用ea:pe(3:1)重结晶。产品过滤,真空干燥得到fmoc-d-arg(pbf)-d-arg(pbf)-oh:37.1g,hplc:98.9%,收率:70.2%。
[0043]
实施例4:fmoc-d-arg(pbf)-d-arg(pbf)-oh的合成
[0044]
取200ml dcm溶解fmoc-d-arg(pbf)-oh(32.4g,50mmol)、honb(9.84g,55mmol),冰水浴的条件下加入逐渐滴加dic(8.5ml,55mmol),然后撤去冰浴,室温搅拌5h。tlc显示原料基本反应完全(pe:ea:hoac=1:1:0.05)反应混合物过滤,滤液备用。
[0045]
将h-d-arg(pbf)-oh(21.32g,50mmol)加入上述滤液中,开始磁力搅拌,并用滴液漏斗滴加dipea(10.74ml,65mmol),室温搅拌10h。反应液浓缩,加入200ml去离子水稀释残留液,ea提取(250*3ml),水层1mol/l盐酸溶液酸化至ph约3,ea萃取,合并ea层,ea层依次用200ml 1mol/l盐酸溶液、200ml*2ml去离子水洗涤2次、200ml用饱和食盐水洗涤1次,用40g无水硫酸钠快速干燥,过滤浓缩,浓缩液用etoh/h2o(1:4)重结晶。产品过滤,真空干燥得到fmoc-d-arg(pbf)-d-arg(pbf)-oh:39.8g,hpl c:99.1%,收率:75.3%。
[0046]
实施例5:fmoc-d-ala-d-arg(pbf)-dpm-nh2的合成
[0047]
称取液相载体dpm-nh2(8.3g,10.0mmol)加入250ml三口烧瓶内,向反应瓶中加入chcl3(80ml),再依次加入hobt(1.3g,10.0mmol)、以及实施2中fmoc-d-ala-d-arg(pbf)-oh(8.64g,12.0mmol)。搅拌溶清。加入edci(2.6g,13mmol),在室温条件下继续搅拌3小时。tlc(dcm:meoh:hac=100:1:0.5)监控反应。反应完全后,反应液在30℃减条件下,减压浓缩至成粘稠物,向粘稠物中加入甲醇(80ml),搅拌2小时。过滤,滤饼用甲醇(30ml
×
3)冲洗三次。将滤饼在40℃条件下真空干燥5小时,得到fmoc-d-ala-d-arg(pbf)-dpm-nh2(14.64g,收率95.5%)。
[0048]
实施例6:fmoc-d-arg(pbf)-d-arg(pbf)-d-ala-d-arg(pbf)-dpm-nh2的合成
[0049]
称量实施例5中化合物fmoc-d-ala-d-arg(pbf)-dpm-nh2(12.27g,8.01mmol)加入1.0l三口烧瓶内,向反应瓶中加入氯仿(400ml),搅拌溶清,再加入dbu(1.22g,8.01mmol)。将反应液冰浴冷却至5℃以下,缓慢滴加二乙胺(6.56g,90mmol),控温不超过5℃。滴加完后,将反应液升至室温继续搅拌2小时。tlc(dcm:meoh:hac=100:1:0.5)监控反应。反应完
全后,将反应液在30℃条件下减压浓缩至成粘稠物,向粘稠物中加入乙腈(80ml),搅拌30分钟。过滤,滤饼用甲醇(40ml
×
2)冲洗两次。将滤饼在40℃条件下真空干燥2小时,得到固体h-d-arg(pbf)-d-arg(pbf)-d-ala-d-arg(pbf)-dpm-nh2=14.67g。
[0050]
将上述固体加入500ml三口烧瓶内,向反应瓶中加入氯仿(150ml),再依次加入ho bt(1.08g,8.01mmol)、以及实施3中fmoc-d-arg(pbf)-d-arg(pbf)-oh(10.15g,9.61mm ol)。搅拌溶清。将反应液冷却至0℃。加入edci(1.69g,8.95mmol),在0-10℃条件下继续搅拌3小时。tlc(dcm:meoh:hac=100:1:0.5)监控反应。反应完全后,反应液在30℃减条件下,减压浓缩至成粘稠物,向粘稠物中加入甲醇(40ml),搅拌2小时。过滤,滤饼用甲醇(40ml
×
3)冲洗三次。将滤饼在40℃条件下真空干燥3小时,得到化合物fmoc-d-arg(pbf)-d-arg(pbf)-d-ala-d-arg(pbf)-dpm-nh2(17.25g,收率91.6%)。
[0051]
实施例7:fmoc-d-ala-d-arg(pbf)-d-arg(pbf)-d-arg(pbf)-d-ala-d-arg(pbf)-dpm-nh2的合成
[0052]
称量实施例6中化合物fmoc-d-arg(pbf)-d-arg(pbf)-d-ala-d-arg(pbf)-dpm-nh2(16.45g,7.0mmol)加入1.0l三口烧瓶内,向反应瓶中加入氯仿(200ml),搅拌溶清,再加入dbu(1.06g,7.0mmol)。将反应液冰浴冷却至5℃以下,缓慢滴加二乙胺(5.47g,75mmol),控温不超过5℃。滴加完后,将反应液升至室温继续搅拌2小时。tlc(dcm:meo h:hac=100:1:0.5)监控反应。反应完全后,将反应液在30℃条件下减压浓缩至成粘稠物,向粘稠物中加入乙腈(70ml),搅拌30分钟。过滤,滤饼用甲醇(40ml
×
2)冲洗两次。将滤饼在40℃条件下真空干燥2小时,得到固体h-d-ala-d-arg(pbf)-d-arg(pbf)-d-arg(pbf)-d-ala-d-arg(pbf)-dpm-nh2=15.95g。
[0053]
将上述固体加入500ml三口烧瓶内,向反应瓶中加入氯仿(160ml),再依次加入ho bt(0.94g,7.0mmol)、以及实施3中fmoc-d-ala-d-arg(pbf)-oh(16.04g,8.4mmol)。搅拌溶清。将反应液冷却至0℃。加入edci(1.45g,7.7mmol),在0-10℃条件下继续搅拌3小时。tlc(dcm:meoh:hac=100:1:0.5)监控反应。反应完全后,反应液在30℃减条件下,减压浓缩至成粘稠物,向粘稠物中加入甲醇(40ml),搅拌2小时。过滤,滤饼用甲醇(40ml
×
3)冲洗三次。将滤饼在40℃条件下真空干燥3小时,得到化合物fmoc-d-ala-d-arg(pbf)-d-arg(pbf)-d-arg(pbf)-d-ala-d-arg(pbf)-dpm-nh2(18.52g,收率93.5%)。
[0054]
实施例8:n-ac-d-cys(mmt)-d-ala-d-arg(pbf)-d-arg(pbf)-d-arg(pbf)-d-ala-d-arg(pbf)-dpm-nh2的合成
[0055]
称量实施例7中化合物fmoc-d-ala-d-arg(pbf)-d-arg(pbf)-d-arg(pbf)-d-ala-d-arg(pbf)-dpm-nh2(18.4g,6.5mmol)加入1.0l三口烧瓶内,向反应瓶中加入氯仿(200ml),搅拌溶清,再加入dbu(0.98g,6.5mmol)。将反应液冰浴冷却至5℃以下,缓慢滴加二乙胺(5.10g,70mmol),控温不超过5℃。滴加完后,将反应液升至室温继续搅拌2小时。tlc(dcm:meoh:hac=100:1:0.5)监控反应。反应完全后,将反应液在30℃条件下减压浓缩至成粘稠物,向粘稠物中加入乙腈(80ml),搅拌30分钟。过滤,滤饼用甲醇(50ml
×
2)冲洗两次。将滤饼在40℃条件下真空干燥2小时,得到固体h-d-ala-d-arg(pbf)-d-arg(pbf)-d-arg(pbf)-d-ala-d-arg(pbf)-dpm-nh2=18.16g。
[0056]
将上述固体加入500ml三口烧瓶内,向反应瓶中加入氯仿(200ml),再依次加入ho bt(0.87g,6.5mmol)、n-ac-d-cys(mmt)-oh(3.39g,7.8mmol)。搅拌溶清。将反应液冷却至0
℃。加入edci(1.35g,7.2mmol),在0-10℃条件下继续搅拌3小时。tlc(dc m:meoh:hac=100:1:0.5)监控反应。反应完全后,反应液在30℃减条件下,减压浓缩至成粘稠物,向粘稠物中加入甲醇(50ml),搅拌2小时。过滤,滤饼用甲醇(50ml
×
3)冲洗三次。将滤饼在40℃条件下真空干燥3小时,得到化合物n-ac-d-cys(mmt)-d-ala-d-arg(pbf)-d-arg(pbf)-d-arg(pbf)-d-ala-d-arg(pbf)-dpm-nh2(18.15g,收率92.3%)。
[0057]
实施例9:n-ac-d-cys(sh)-d-ala-d-arg(pbf)-d-arg(pbf)-d-arg(pbf)-d-ala-d-arg(pbf)-dp m-nh2的合成
[0058]
称量实施例8中化合物n-ac-d-cys(mmt)-d-ala-d-arg(pbf)-d-arg(pbf)-d-arg(pbf)-d-al a-d-arg(pbf)-dpm-nh2(18.0g,5.95mmol)加入1.0l三口烧瓶内,加入300ml 1%tfa/chcl3,室温磁力搅拌5分钟后,反应液在30℃条件下减压浓缩至成粘稠物,向粘稠物中加入乙腈(80ml),搅拌30分钟。过滤,滤饼用甲醇(50ml
×
2)冲洗两次;重复该过程2次,一共3次。得到化合物n-ac-d-cys(sh)-d-ala-d-arg(pbf)-d-arg(pbf)-d-arg(pbf)-d-ala-d-arg(pbf)-dpm-nh2(17.65g,收率98.05%)
[0059]
实施例10:c
152h243n21o26
s6的合成
[0060]
称量实施例9中化合物n-ac-d-cys(sh)-d-ala-d-arg(pbf)-d-arg(pbf)-d-arg(pbf)-d-ala-d-arg(pbf)-dpm-nh2(17.65g,6.41mmol)加入1.0l三口烧瓶内,向反应瓶中加入氯仿(250ml),再boc-cys(npys)-oh(2.64g,7.05mmol)。搅拌溶清。将反应液冷却至0℃。加入dipea(2.0ml,10mmol),在20-30℃条件下继续搅拌24小时。tlc(dcm:meoh:ha c=100:1:0.5)监控反应。反应完全后,反应液在30℃减条件下,减压浓缩至成粘稠物,向粘稠物中加入甲醇(50ml),搅拌2小时。过滤,滤饼用甲醇(50ml
×
3)冲洗三次。将滤饼在40℃条件下真空干燥3小时,得到化合物c
152h243n21o26
s616.19 g,收率85%,其结构式如下。
[0061][0062]
实施例11:c
38h73n21o10
s2的合成
[0063]
将实施例10中的化合物16.0g置于1.0l反应瓶中,以10ml/g化合物的比例加入160ml裂解试剂tfa:h2o:phsme:anisole:tis=88:5:3:2:2,室温磁力搅拌进行裂解反应2.5小时。然后将反应液缓慢倒入冰冻mtbe(1600ml)中,搅拌30分钟后,冰箱中静置1小时。离心,用乙醚(100ml
×
3)洗涤三次。得到的沉降物在30℃条件下干燥2小时,再用甲醇(100ml)打浆2小时,过滤,滤饼弃去,滤液40℃旋干后得到粗肽(10.03g,136.8%),即是etelcalcetide(化学式c
38h73n21o10
s2)。粗肽hplc纯度约为89.34%。
[0064]
经过纯化,得到etelcalcetide精肽6.08g,总收率为82.95%,精肽的纯度为99.37%。其化学结构式如下:
[0065][0066]
上述实施例1-11中涉及的缩写及英文含义如下表1所示。
[0067]
表1缩写及英文含义
[0068]
[0069]
[0070]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1