一种合成核苷及其类似物的方法和试剂与流程
本发明涉及核苷及其类似物的合成。更具体地说,本发明涉及一种合成核苷及其类似物的方法和试剂。
背景技术:
核苷在从细胞信号传导到代谢的各种细胞过程中发挥关键作用(1)。dna(25)和rna(26)的益生元合成被提议涉及核碱基型烯胺和甘油醛之间的偶联,以“核糖末梢”方法在呋喃糖之前形成核碱基亚氨基离子。合成核苷类似物(nas),旨在模拟它们的天然对应物,被广泛地在药物化学中应用,并在化学生物学中被用作工具化合物(2-18)。nas已被用于癌症的治疗(2,6),是一类最大的小分子抗病毒药物(3,4),在机制上,nas可以作为有毒的抗代谢物,干扰核酸的合成(4),另外,在体内磷酸化之后,所产生的核苷酸类似物可以抑制参与癌细胞生长或病毒复制的酶(例如,dna/rna聚合酶,核糖核苷酸还原酶或核苷磷酸化酶)(2、4)。nas也显示出了作为表观遗传调节剂的前景,地西他滨和氮杂胞苷都能抑制dna甲基转移酶,并已被批准用于癌症治疗(4)。然而,nas的合成过程往往是漫长的,不适合多样化,并依赖于有限的手性碳水化合物原料库,因此存在许多挑战(例如,19-24、27、33、42-44)。锁定核酸(lnas)(39)是一种构象受限的nas,表现出更好的稳定性,其与反义寡核苷酸的结合可显著提高特异性和效力。然而,与其他c4’修饰的nas的合成非常相似,lna的合成通常是长期的。
技术实现要素:
本发明涉及核苷及其类似物的合成。在一方面,本发明提供了一种合成核苷或其类似物的方法,所述方法通过脯氨酸催化对芳基或杂芳基取代的乙醛化合物进行卤化,然后进行对映选择性羟醛缩合反应,生成卤代醇化合物;还原卤代醇化合物生成卤代醇二醇(halohydrin diol)化合物;以及在环化卤化物置换(ahd)反应中使卤代醇二醇化合物与路易斯酸或碱接触,生成核苷或其类似物。在一些实施例中,所述路易斯酸可以是incl3或sc(otf)3。在一些实施例中,所述卤代醇二醇化合物可以在路易斯碱处理之前分离。在一些实施例中,所述碱可以是naoh。在一些实施例中,所述碱-ahd反应可以产生c3'、c5'保护的核苷或其类似物。在可替代方面,本发明提供了一种在合成核苷或其类似物中制备中间体的方法:通过脯氨酸催化对杂芳基取代的乙醛化合物进行卤化,然后进行对映选择性羟醛缩合反应生成卤代醇化合物;还原卤代醇化合物获得卤代醇二醇化合物,以在合成核苷或其类似物中生成中间体。
在可替代方面,本发明提供了一种合成核苷或其类似物的方法:(i)提供卤代醇二醇化合物;ii)在环化卤化物置换(ahd)反应中使卤代醇二醇化合物与路易斯酸或碱接触,以生成核苷或其类似物。本发明的发明内容并不一定描述本发明的所有特征。
附图说明
本发明的这些和其他特征将从以下描述中变得更加明显,其中参考附图包括:图1是通过一系列反应合成核苷和核苷类似物(nas)的示意图,这些反应包括不对称α-氟化羟醛缩合反应(αfar),然后是环化(环化annulation)反应包括环氟置换(afd反应)。het=杂芳基。图2a-c显示了吡唑烷基na 17的合成。a:核苷的益生元合成被认为涉及以“核糖末梢”的方法将核苷烯胺如12与甘油醛偶联。一种合成核糖末梢nas的方法包括亚胺离子替代物14的羟醇缩合反应。b:对脯氨酸催化的α-氟化和羟醇缩合反应的检查表明,该方法与α-吡唑醛15相容,以良好的产率和对映选择性提供氟醇16。减少和环化氟化物置换(afd)为na 17提供了快速途径。c:机理研究表明,afd通过立体化学转化(sn2反应)进行的,然后进行差向异构。nfsi=n-氟苯磺酰亚胺;dmf=二甲基甲酰胺;mecn=乙腈;otf=三氟甲磺酸酯。图3a-f显示了核苷和na的合成。a:一个4步的反应序列将现成的起始材料转化为对映体富集和自然配置的β-d-nas。b:naoh可促进afd产生尿嘧啶、胸腺嘧啶、吡唑基和5-嘧啶基核苷和nas。c:afd产生三氟甲基尿嘧啶、三唑基、邻苯二甲酰亚胺、脱氮腺嘌呤、腺苷核苷和nas可由路易斯酸sc(otf)3或incl3促进。d:nas在c3’和c5
’‑
醇功能上都有保护作用。e:非天然核苷(l-对映体)利用d-脯氨酸催化αfar反应。f:c2’修饰的nas。atempo,baib,二恶烷(92%来自34)。bi)硫代羰基二咪唑(thiocarbonyldiimidazole),thf;ii)bu3snh,偶氮二异丁腈(从35起2步55%)。ci)tempo,baib,二恶烷;ii)memgbr,thf,-78℃(从34起2步80%)。ddast,ch2cl2,然后是hcl,meoh(53%来自35)。tempo=2,2,6,6-四甲基哌啶-1-基)氧基;baib=双(乙酰氧基)碘苯(bis(acetoxy)iodobenzene);thf=四氢呋喃;dast=三氟化二乙氨基硫(diethylaminosulfur trifluoride)。图4a-e显示了c4’修饰的和其他nas的快速合成。a:将有机镁试剂添加到αfar产物中生成叔醇,直接进行afd或lewis酸/碱促进afd到c4’修饰的nas。b:氟醇55的大规模(~380g)生产支持mk-3682(hcv rna聚合酶抑制剂)的合成。c:氟醇59的还原胺化提供了亚胺基核苷60的直接途径。d:利用c3’和c5
’‑
oh功能的固有保护作用,制备c4’修饰的c2
’‑
脱氧na 62。e:两种lna 65和68的合成。a酮-氟醇羟醛缩合加合物的收率。b非对映体的联合产量。c用csa和二甲氧基丙酮将粗反应混合物加热到50℃后的产物。d用hcl水溶液处理粗反应混合物后的产物。e从单一的氟醇59开始。
具体实施方式
详细说明本发明公开部分提供了合成核苷或其类似物的方法和中间体。图1显示了使用简单的非手性合成砌块通过脯氨酸催化的α-氟化和羟醇缩合反应(α-far)和环氟置换(afd),合成核苷类似物(na)。所述合成包括一步法通过脯氨酸催化杂
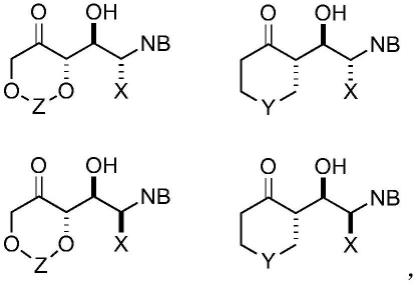
芳基取代的乙醛9进行α-氟化-羟醛缩合反应,然后通过还原或有机金属加成和afd反应。例如,该过程允许直接获得c3'/c5'保护的na 10(和c2'修饰的na),提供该碱基取代的灵活性,提供通向c4'修饰的na的直接途径等。在一些实施例中,该方法包括互补(核糖末梢(ribose-last))方法,该方法还涉及碱基-亚胺离子的末端环化,用于合成核苷和na的合成。在一种被提议的dna益生元(prebiotic)的合成中,碱基型烯胺11(图2a)和甘油醛之间的偶联在呋喃糖之前以“核糖末梢”的方式形成一个碱基亚胺离子12。作为碱基亚胺离子12的合成等价物,提出了卤代无环na 13(图2a)。在不受任何特定理论约束的情况下,通过二羟基丙酮衍生物(例如,8)(30)和α-卤代醛14(图2a)的有机催化羟醛缩合反应,可以形成核糖核苷c2'-c3'键并控制相对和绝对立体化学。因此,本发明描述的方法包括i)利用已知不稳定的卤代醛(如28、29、31、32、35)的反应性,与连接在同一位置的碱基结合(例如,8)和ii)在最后一步中发展环化卤化物置换(ahd)反应以形成核糖环。在一些实施例中,本发明提供了一种合成核苷和na的方法,使用简单的非手性材料,通过一个短的(2-3步)序列反应,包括“一步法”脯氨酸催化杂芳基取代的乙醛的α-卤代反应以及串联对映选择性羟醛缩合反应(αhar),然后进行还原或有机金属加成和环化(环化)反应,包括环化(环化annulation)卤化物置换(ahd)反应。更具体地说,在一些实施例中,本发明提供了一种合成核苷或其类似物的方法,通过:(i)通过脯氨酸催化将芳基或杂芳基取代的乙醛化合物进行卤化,生成α-卤醛化合物,然后通过脯氨酸催化与酮偶联生成卤代醇化合物。(ii)还原卤代醇化合物生成卤代醇二醇化合物;和(iii)在环化卤化物置换(ahd)反应中使卤代醇二醇化合物与路易斯酸或碱接触,生成核苷或其类似物。在一些实施例中,所述路易斯酸可以是但不限于是嗜盐路易斯酸。在一些实施例中,所述路易斯酸可以是但不限于是incl3或sc(otf)3。在一些实施例中,路易斯酸-促进的ahd可产生c2',c3'保护的核苷或na。在一些实施例中,lewis酸-促进的ahd可能导致保护基团迁移,即可能产生具有迁移的缩丙酮保护基团的na。在一些实施例中,路易斯酸-促进的ahd可能导致脱保护。在一些实施例中,所述碱可以是naoh。在一些实施例中,所述碱基-促进的ahd可以产生c3',c5'-保护的na。在一些实施例中,在路易斯碱处理之前,αhar反应产物可以被还原和分离。在一些实施例中,本发明提供了一种在合成核苷或核苷类似物中制备中间体的方法,通过:(i)通过脯氨酸催化对杂芳基取代的乙醛化合物进行卤化,然后进行对映选择性羟醛缩合反应生成卤代醇化合物;(ii)然后还原卤代醇化合物以得到卤代醇二醇化合物,在合成核苷或其类似物时生成中间体。在一些实施例中,本发明提供了一种合成核苷或其类似物的方法,通过:
(i)提供卤代醇二醇化合物;以及ii)在环化卤化物置换(ahd)反应中使卤代醇二醇化合物与路易斯酸或碱接触,生成核苷或其类似物。“卤代醇”是指一种含有官能团的化合物,其中一个卤素和一个羟基与相邻的基团相连。卤代醇可以有以下一般结构,其中r1、r2可以是任何合适的基团,如本发明所示,x也如本发明所示:在一些实施例中,所述卤代醇化合物可以具有以下一般结构,其中nb和x可以如本发明所示:在一些实施例中,所述卤代醇化合物可以被芳基或杂芳基功能化,即,nb可以是芳基或杂芳基。在一些实施例中,所述卤代醇二醇化合物可具有以下一般结构,其中nb和x可如本发明所示:在一些实施例中,所述卤代醇二醇化合物可以用芳基或杂芳基功能化,即nb可以是芳基或杂芳基。在一些实施例中,本发明公开提供以下核苷或其类似物,包括但不限于其非对映体,其中nb可以如本发明所示,每个r可独立为-oh、-oc(ch3)2o-、-(ch2)
3-、-ch2sch
2-或-ch2och
2-:在一些实施例中,本发明公开提供以下化合物或其对映体,其中nb和x可以如本发明所示,并且每个r可以独立地是-oh、-oc(ch3)2o-,-(ch2)
3-,-ch2sch
2-,或-ch2och
2-,用于合成核苷或其类似物的中间体:
在一些实施例中,本发明提供了以下化合物或其对映体,其中nb和x可以如本发明所示,y可以是ch2、o、s、nr,其中r可以是烷基或芳基,z可以是乙醇的保护基团,包括但不限于缩丙酮(acetonide)、甲硅烷基保护基、烷基保护基或芳基保护基(包括环基或非环基),用于合成核苷或其类似物的中间体:在一些实施例中,本发明提供以下化合物或其对映体,其中nb和x可以如本发明所示,用于合成核苷或其类似物的中间体:在一些实施例中,本发明公开提供了以下化合物或其对映体,其中nb和x可以如本发明所示,而y可以是ch2、o、s、nr,其中r可以是烷基或芳基,用于合成核苷或其类似物的中间体:在一些实施例中,本发明公开的方法以良好的对映选择性和/或产率(例如,大于10g至约400g,或介于10g、15g、20g、25g、50g、75g、100g、125g、150g、200g、250g、300g、350g或400g之间的任何值)快速获得合成核苷或其类似物的中间体。因此,本发明所公开的方法可用于核苷和/或nas的工艺规模生产。在一些实施例中,本发明公开的方法能够直接获得c3'/c5'保护的na3,其中r可以是烷基、炔基或芳基,nb可以如本发明所示(和因此c2'修饰的nas),提供碱基取代的灵活
benzodioxanyl)等。除非在此特别说明,术语“芳基”是指包括任选地由本发明所述的一个或多个取代基取代的芳基。“杂芳基”是指在环中含有一个或多个杂原子(例如n、o、s)的单个或稠合芳香环基团,包括例如5-14个成员,如5、6、7、8、9、10、11、12、13或14个成员。杂芳基的例子包括呋喃、噻吩、吡咯、噁唑、噻唑、咪唑、吡唑、异噁唑、异噻唑、1,2,3-噁二唑、三唑(例如,1,2,3-三唑或1,2,4-三唑),1,3,4-噻二唑,四唑,吡唑,吡啶,哒嗪,嘧啶,2,6-二氯嘧啶吡嗪,1,3,5-三嗪,咪唑,苯并咪唑,苯并噁唑,苯并噻唑,吲哚嗪,吲哚,异吲哚,苯并呋喃、苯并噻吩、1h-吲唑、嘌呤、4h-喹嗪(4h-quinolizine)、喹啉、异喹啉、噌啉、酞嗪、喹唑啉、喹喔啉、1,8-萘啶、蝶啶、尿嘧啶、胸腺嘧啶、脱氮腺嘧啶(deazadenine)、邻苯二甲亚酰胺、腺嘌呤等。除非本发明另有具体说明,术语“杂芳基”是指包括被本发明所述的一个或多个取代基选择性取代的杂芳基。卤素包括溴、氯、氟、碘等,在本发明公开的化学结构中用“x”表示。在一些实施例中,卤素可包括氯或氟。据此,“卤素(halo)”指的是溴、氯、氟、碘等。卤化物是带有负电荷的卤素原子。所谓“卤化”是指将卤素原子引入化合物或分子中。“可选的”或“可选地”是指随后描述的事件或情况可能发生或不发生,并且描述包括该事件或情况发生一次或多次的情况和不发生的情况。例如,“任选取代的烷基”意指烷基可以被取代,也可以不被取代,描述中包括被取代的烷基和没有取代的烷基,并且烷基可以被取代一次或多次。任选取代的烷基的例子包括但不限于甲基、乙基、丙基、丁基、戊基、己基、异丙基、异丁基、仲丁基、叔丁基等。合适的可选取代基的例子包括但不限于h,f,cl,ch3,oh,och3,cf3,chf2,ch2f,cn,卤素,和c
1-10
烷氧基。除非上下文另有明确规定,否则这里使用的单数形式“a”、“and”和“the”包括复数指代。例如,“一种化合物”指的是这种化合物中的一种或多种。在本发明中,术语“化合物compound”或“化合物compounds”是指本发明所讨论的化合物,包括这些化合物的前体和衍生物。本发明的化合物可包含一个或多个不对称中心,因此可以外消旋体和外消旋混合物、单一对映体、非对映体混合物和单个非对映体的形式出现。根据分子上各种取代基的性质,还可能存在其他不对称中心。每个这样的不对称中心将独立地产生两种光学异构体,并且打算将所有可能的光学异构体和非对映体的混合物以及纯的或部分纯化的化合物都包括在本发明的范围内。本说明书中描述的化合物的任何公式、结构或名称,如果没有指定特定的立体化学,则意味着包括上述的任何和所有现有的异构体以及其任何比例的混合物。当指定立体化学时,本发明意在包括纯形式的特定异构体或与其他异构体以任何比例的混合物的一部分。单一对映体,即光学活性形式,可以通过不对称合成或通过外消旋体的拆分获得。外消旋体的拆分可以通过常规方法完成,例如在有拆分剂的情况下进行结晶;使用例如手性高效液相色谱柱进行色谱;或者用拆分剂衍生外消旋体混合物以产生非对映体,通过色谱分离非对映体,并去除拆分剂以产生对映体富集的原始化合物。如果需要,可以重复这些步骤,以提高化合物的对映体纯度。当本发明所述的化合物含有油性(olefmic)双键或其他几何不对称中心时,除非另有规定,否则这些化合物应包括顺式、反式、z-和e-构型。同样,所有的同分异构体形式也被包括在内。本发明所述起始材料可以从商业来源获得,由商业可用的有机化合物制备,使用已知的合成方法制备。
本发明将在以下实施例中进一步说明。实施例材料与方法常规考虑因素l-和d-脯氨酸(纯度99%)购自alfa aesar。除非另有说明,所有描述的反应都在环境温度和大气下进行。使用230-400目硅胶(e.merck,silica gel 60)进行柱色谱。微量溶剂的浓缩和去除是通过使用丙酮-干冰冷凝器和welch真空泵的buchi旋转蒸发器完成的。使用氘代氯仿(cdcl3)、氘代甲醇(cd3od)、氘代丙酮((cd3)2co)、氘代乙腈(cd3cn)或氘代二甲基亚砜(dmso-d6)作为溶剂记录核磁共振(nmr)光谱。信号位置(δ)以百万分之四甲基硅烷(δ0)为单位给出,相对于溶剂的信号进行测量(1h nmr:cdcl3:δ7.26;cd3od:δ3.31;(cd3)2co:δ2.05;cd3cn:δ1.96;dmso-d6:δ2.50;
13
c nmr:cdcl3:δ77.16;cd3od:δ49.00;(cd3)2co:δ29.84;cd3cn:δ1.32;dmso-d6:39.5)。耦合常数(j值)以赫兹(hz)为单位,并报告为最接近的0.1hz。1h nmr光谱数据按以下顺序列出:多重性(s,单线;d,双线;t,三线;q,四线;sept,七线;m,多线;br宽)、耦合常数、质子数。在配备有qnp或tci冷冻探针(600mhz)、bruker 400(400mhz)或bruker 500(500mhz)的bruker avance 600上记录nmr光谱。非对映体比率(dr)是基于对粗品的1h nmr的分析。1h的分配是基于对1h-1
h-cosy和noe光谱的分析。
13
c的分配是基于hsqc光谱的分析。高效液相色谱(hplc)分析是在agilent 1100hplc上进行的,配备了一个可变波长的uv-vis检测器。在perkin elmer spectrum two ftir光谱仪上清晰地记录了红外(ir)光谱。仅为每种化合物提供选定的特征吸收数据。用perkin-elmer 341偏振仪在589nm处测量旋光率。常规步骤常规步骤a(一锅式有机催化α-氟化/羟醛缩合反应)在4℃下,向nfsi(1.5当量)、l-脯氨酸(1.5当量)和nahco3(1.5当量)在dmf(0.75m)中的搅拌悬浮液中加入醛(1.5当量)样品。当通过1hnmr光谱分析观察到完全转化为α-氟甲醛(α-fluoroaldehyde)时,然后加入2,2-二甲基-1,3-二恶烷-5-酮(8)(1.0当量)的ch2cl2或thf或mecn(1.25*dmf体积)溶液,并将所得混合物加热至室温。36-72小时后,或通过对小份反应物的1h nmr光谱分析观察到8完全反应时,用ch2cl2稀释该混合物,并用饱和碳酸氢钠溶液和水各洗一次有机层。然后通过mgso4干燥有机层,减压浓缩,并按规定用闪蒸色谱法提纯粗产物。常规步骤b(同步-还原)(syn-reduction)在-15℃下,向顺式-和反式-氟醇(fluorohydrins)(1.0当量)在mecn(0.10m)的搅拌溶液中加入四甲基三乙酰氧基硼氢化铵(tetramethylammoniumtriacetoxyborohydride)(5.0当量)和乙酸(10当量)。将得到的混合物搅拌16小时或直到起始物料反应完全(通过tlc分析确定)。然后用罗谢尔盐的饱和溶液稀释反应混合物,用ch2cl2洗涤三次。分离有机层,并用mgso4干燥,减压浓缩,并通过闪蒸色谱法提纯粗产物。常规步骤c(碱促进环化)
向顺式-二醇、顺式-和反式氟醇(1.0当量)在mecn(0.10m)的搅拌溶液中加入2mnaoh(2.5-10当量),然后将反应混合物搅拌5小时或直到起始物料反应完全(通过tlc分析确定)。然后反应混合物用ch2cl2稀释并用饱和氯化铵溶液洗涤。分离有机层,并用mgso4干燥,过滤,减压浓缩。通过闪蒸色谱法提纯粗产物。常规步骤d(路易斯酸促进环化)向顺式-二醇、顺式-和反式氟醇(1.0当量)在mecn(0.10m)的搅拌溶液中加入sc(otf)3或incl3(0.10-2.5当量),然后将反应混合物搅拌6小时或直到起始物料反应完全(通过tlc分析确定)。然后将反应混合物用ch2cl2稀释并用饱和碳酸氢钠溶液洗涤。分离有机层,并用mgso4干燥,过滤,减压浓缩。通过闪蒸色谱法提纯粗产物。常规步骤e(格氏加成法)将氟醇羟醛(fluorohydrin aldol)加合物(1当量)在ch2cl2(0.025m)中的搅拌溶液冷却到-78℃。然后滴加有机镁试剂(2.2-5当量),并将所得反应混合物搅拌5小时。然后在-78℃用氯化铵:甲醇溶液(1:1-饱和氯化铵溶液:甲醇)淬灭反应混合物,并加热至室温。将得到的混合物用ch2cl2稀释,并用水清洗两次。有机层用mgso4干燥,过滤,减压浓缩,得到粗产物。然后粗产物通过闪蒸色谱法提纯或直接用于环化。化合物的制备和表征s1、醛sm1、醇醛加合物a1、二醇加合物18a/18b和核苷类似物17、19和34的制备。将吡唑(1.00克,14.7毫摩尔,1.0当量)、溴乙醛二乙基缩醛(bromoacetaldehyde diethyl acetal)(2.67毫升,17.6毫摩尔,1.2当量)和k2co3(4.06克,29.4毫摩尔,2.0当量)的溶液在dmf(74毫升)中于90℃搅拌36小时。然后过滤反应混合物,用40毫升ch2cl2洗涤并减压浓缩。通过闪蒸色谱法(戊烷:乙酸乙酯-7:3)纯化粗产物s1,得到si(2.43克,产率90%),其为无色油。将s1(0.100克,0.543,1.0当量)在0.5m盐酸(0.54毫升)中的溶液加热至90℃,持续5小时。在完全转化为sm1后,将反应混合物减压浓缩,所得产物sm1用于下一步反应,无需提纯。s1的数据:ir(纯):υ=2977,2904,1516,1396,1129,1063,751,621cm-1
;1h nmr(400mhz,cdcl3):δ7.51(d,j=1.8hz,1h),7.46(d,j=2.3hz,1h),6.24(dd,j=2.3,1.8hz,1h),4.77(t,j=5.5hz,2h),4.22(d,j=5.5hz,2h),3.70(m,2h),3.41(m,2h),1.16(t,j=7.1hz,6h);
13
c nmr(125mhz,cdcl3):δ139.7,130.6,105.6,101.7,63.8,55.2,15.3hrms(ei
+
)计算得c9h
17
n2o2[m+h]
+
185.1285;found 185.1284α-氟化/醇醛缩合按照常规步骤a,将sm1(0.543毫摩尔)、nfsi(0.170克,0.543毫摩尔)、l-脯氨酸(0.063克,0.543毫摩尔)和nahco3(0.045克,0.543毫摩尔)的溶液在dmf(0.72毫升)中于4℃搅拌12小时。然后加入8(0.043毫升,0.362毫摩尔)在mecn(0.90毫升)中的溶液,然后将反应混合物在室温下搅拌60小时。通过闪蒸色谱法(戊烷:et2o-25:75)纯化粗产物氟醛a1
(0.060克,产率64%,dr 1.4:1),得到淡黄色油状物的顺式-和反式-氟醛a1。顺式-和反式氟醇a1的数据:ir(neat):υ=2989,1749,1446,1376,1091,1042,764cm-1
;1h nmr(600mhz,cdcl3):δ7.88,7.78,7.63,6.45,6.44,6.39,6.37,4.89,4.50,4.36,4.34,4.31,4.26,4.07,4.04,1.50,1.45,1.45,1.34;
13
c nmr(150mhz,cdcl3):δ209.0,207.4,141.7,141.4,131.5,131.1,107.7,107.5,101.8,101.4,95.0,94.6,74.3,72.4,71.0,70.2,67.0,66.9,24.0,23.7,23.7,23.4;
19
f nmr(470mhz,cdcl3):δ-144.9,-154.1hrms(ei
+
)计算得c
11h16
fn2o4[m+h]
+
259.1089;found 259.1093顺式-和反式-氟醇a1的同步还原按照常规步骤b,在-15℃下,将me4nhb(oac)3(0.968克,3.68毫摩尔)和acoh(0.442毫升,7.36毫摩尔)加入到a1(0.190克,0.736毫摩尔)在mecn(7.36毫升)的搅拌溶液中,并将反应混合物搅拌18小时。通过闪蒸色谱法(戊烷:乙酸乙酯-1:1)纯化粗二醇18a和18b,得到无色油状的18a和18b的混合物(0.151克,产量79%,d.r.(顺/反)=1:1.2)。顺式-二醇、顺式-氟醛18a的数据:[α]
d20
=+83.2(c 0.37in mecn);ir(neat):υ=3001,1442,1375,1039,918,749cm-1
;1h nmr(600mhz,cdcl3):δ7.68(d,j=2.4hz,1h),7.64(d,j=1.5hz,1h),6.38(dd,j=2.4,1.5hz,1h),6.18(d,j=51.2hz,1h),4.27(dd,j=22.4,8.8hz,1h),3.95(dd,j=11.1,5.6hz,1h),3.93(dd,j=9.5,8.0hz,1h),3.80(m,1h),3.70(dd,j=11.2,11.0hz,1h),1.52(s,3h),1.39(s,3h);
13
c nmr(150mhz,cdcl3):δ141.5,132.0,107.2,99.0,91.9(d,j=211.0hz),72.3(d,j=21.8hz),70.6,67.1,63.8,28.7,19.4;
19
f nmr(470mhz,cd3cn):δ-150.3hrms(ei
+
)计算得c
11h18
fn2o4[m+h]
+
261.1245;found 261.1255顺式-二醇、反式-氟醇18b的数据:[α]
d20
=-10.8(c 0.91in mecn);ir(neat):υ=3646,3001,1443,1375,1039,918cm-1
;1h nmr(600mhz,cdcl3):δ7.70(d,j=0.9hz,1h),7.65(d,j=2.5hz,1h),6.40(dd,j=2.5,0.9hz,1h),6.29(dd,j=48.4,2.9hz,1h),4.41(ddd,j=8.0,4.0,2.9hz,1h),3.87(m,2h),3.52(dd,j=11.3,2.7hz,1h),3.17(dd,j=8.8,8.8hz,1h),1.34(s,3h),1.16(s,3h);
13
c nmr(150mhz,cdcl3):δ142.1,132.0,106.9,98.9,93.1(d,j=207.9hz),76.2(d,j=24.7hz),72.2(d,j=5.3hz),67.3(d,j=4.6hz),
63.8,28.5,19.3;
19
f nmr(470mhz,cd3cn):δ-145.9hrms(ei
+
)计算得c
11h18
fn2o4[m+h]
+
261.1245found 261.1262二醇18a和18b的环化按照常规步骤c,二醇18a和18b分别环化为同一产品(17)。由18b的sn2环化产生的α-异构体(α-anomer)在反应条件下环化为热力学上更稳定的β-异构体17。此外,采取2:1的产品混合物(19:17),并按照常规步骤c,只得到β-异构体17。还请注意,17的e.r.(95:5)代表18a(93:7)和18b(98:2)的平均e.r.。按照常规步骤c,将18a和18b(0.025克,0.096毫摩尔,d.r.(syn/anti)=1:1)和2m naoh(0.48毫升,0.962毫摩尔)的混合物在50℃下在mecn(0.96毫升)中搅拌5小时。通过闪蒸色谱法(戊烷:乙酸乙酯-65:35)纯化粗产物34,得到白色固体核苷类似物34(0.018克,收率76%)。有时可以观察到高达5:1(β:α)的产物混合物。核苷类似物34的数据:[α]
d20
=-58.9(c 2.0in mecn);ir(neat):υ=3339,2926,1647,1450,1397,1092,1045,759cm-1
;1h nmr(400mhz,cd3cn):δ7.70(d,j=2.4hz,1h),7.56(d,j=1.6hz,1h),6.30(dd,j=2.4,1.6hz,1h),5.70(s,1h),4.47(d,j=4.6hz,1h),4.12(dd,j=9.6,4.6hz,1h),4.11(dd,j=9.6,4.6hz,1h),3.91(dd,j=10.3,9.6hz,1h),3.83(dd,j=9.6,4.6hz,1h),3.72(br s,1h),1.54(s,3h),1.43(s,3h);
13
c nmr(100mhz,cd3cn):δ141.7,130.1,106.7,101.7,96.1,74.7,74.4,71.8,65.9,29.3,20.1hrms(ei
+
)calcd for c
11h17
n2o4[m+h]
+
241.1183;found 241.1197核苷类似物34的脱保护将34(0.021克,0.088毫摩尔)溶于meod(1.0毫升)中,并加入两滴1m hcl,然后将溶液在室温下放置12小时。随后,在减压下浓缩反应混合物,得到白色固体17(0.018克,
100%)。核苷类似物17的数据:[α]
d20
=+70.4(c 0.48in meoh);ir(neat):υ=3325,2944,2832,1449,1022,631cm-1
;1h nmr(600mhz,cd3cn):δ7.74(d,j=2.3hz,1h),7.58(d,j=1.0hz,1h),6.30(dd,j=2.3,1.0hz,1h),5.70(d,j=4.3hz,1h),4.51(m,1h),4.33(m,1h),4.08(br s,1h),3.74(dd,j=12.3,2.8hz,1h),3.67(d,j=5.7hz,1h),3.59(dd,j=12.3,2.5hz,1h),3.52(d,j=4.3hz,1h);
13
c nmr(150mhz,cd3cn):δ141.2,131.1,106.4,94.7,87.2,76.6,72.3,63.4.hrms(ei
+
)计算得c8h
13
n2o4[m+h]
+
201.0870;found 201.0870二醇18b的环化将18b(0.043克,0.165毫摩尔)和2m naoh(0.21毫升,0.443毫摩尔,2.5当量)的溶液在50℃下在mecn(1.65毫升)中搅拌3小时。通过闪蒸色谱法(戊烷:乙酸乙酯-65:35)提纯粗产物19,得到白色固体核苷类似物19(0.026克,收率76%)。核苷类似物19的数据:[α]
d20
=+72.2(c 0.98in mecn);ir(纯):υ=3366,2992,1306,1383,1200,1076,754cm-1
,1h nmr(600mhz,cd3cn):δ7.76(d,j=2.3hz,1h),7.56(d,j=1.2hz,1h),6.35(d,j=2.3hz,1h),5.38(d,j=0.9hz,1h),4.12(dd,j=0.9,2.1hz,1h),3.94(d,j=2.1,9.7hz,1h),3.81(dd,j=5.0,10.6hz,1h),3.59(m,2h),3.37(m),1.45(s,3h),1.33(s,3h);
13
c nmr(150mhz,cdcl3):δ142.1,131.0,108.2,99.9,71.8,65.4,65.2,64.7,59.0,29.1,19.9.hrms(ei
+
)计算得c
11h17
n2o4[m+h]
+
241.1183;found 241.1176二醇18a的相对立体化学的测定二醇18a被转化为双-对-硝基-苯甲酰酯(bis-p-nitro-benzoyl ester)并在乙醇中重新结晶。这样就可以用单x射线晶体学来确定相对立体化学。核苷类似物17的相对立体化学的测定核苷类似物17的2d noesy分析支持指定的立体化学。核苷类似物19的相对立体化学的测定
核苷类似物19的2d noesy分析支持指定的立体化学。二醇18a的对映体过量的测定按照常规步骤a和b,使用1:1的l-:d-脯氨酸混合物,制备二醇18a的外消旋品。使用3μm amylose-1色谱柱通过手性hplc分离对映体二醇,流速0.40ml/min;洗脱剂:己烷-iproh 90:10;检测波长210nm;保留时间:(+)-18a为6.66min;(-)-18a为8.10min。使用相同的方法(93:7e.r)测定光学富集的(+)-18a二醇的对映体比率。二醇18b的对映体过量的测定按照常规步骤a和b,使用1:1的l-:d-脯氨酸混合物,制备二醇18b的外消旋品。使用3μm amylose-1色谱柱通过手性hplc分离对映体二醇,流速0.40ml/min;洗脱剂:己烷-iproh 90:10;检测波长210nm;保留时间:(-)-18b为6.13min;(+)-18b为11.72min。使用相同方法(98:2e.r)测定光学富集的(-)-18b二醇的对映体比率。核苷类似物34的对映体过量的测定按照常规步骤a、b和c,使用1:1的l-:d-脯氨酸混合物,制备核苷34的外消旋品。使用3μm-i-纤维素-5柱通过手性hplc分离核苷对映体;流速0.10ml/min;洗脱剂:己烷-iproh 90:10;检测波长254nm;保留时间:(-)-34为8.91min;(+)-34为13.32min。使用相同方法(95:5e.r)测定光学富集的(-)-34的对映体比率。羟醛缩合加合物a2、二醇加合物d2和核苷类似物24、35和ent-24的制备α-氟化/羟醛缩合按照文献步骤(45)制备相应的起始醛/水合物sm3。按照常规步骤a,将醛(1.32mmol)、nfsi(0.416g,1.32mmol)、l-脯氨酸(0.152g,1.32mmol)和nahco3(0.111g,1.32mmol)的溶液在4℃下在dmf(1.76ml)中搅拌12小时。然后加入8(0.105毫升,0.880毫摩尔)在thf(2.64毫升)中的溶液,反应混合物在4℃下搅拌96小时。通过闪蒸色谱法(戊烷:乙酸乙酯-1:1)纯化粗制的氟醛a2,得到不可分离的淡白色固体混合物顺式-和反式氟醛a2(0.159克,产率60%,d.r.1.2:1)。顺式-和反式氟醛a2的数据:ir(neat):υ=3432,2992,2900,1692,1381,1079cm-1
;1h nmr(600mhz,cdcl3):δ8.87,8.79,7.74,7.68,6.68,6.67,5.80,5.77,4.53,4.40,4.34,4.33,4.30,4.13,4.11,4.06,3.70,3.48,1.52,1.46,1.44,1.44;
13
c nmr(150mhz,cdcl3):δ211.3,208.7,162.8,162.6,150.3,149.8,141.7,141.1,103.2,102.6,102.1,101.9,90.7,90.3,73.3,71.4,70.7,70.5,66.6,66.5,23.7,23.6,23.6,23.3;
19
f nmr(470mhz,cdcl3):
δ
–
162.0,
–
178.6.hrms(ei
+
)计算得c
12h16
fn2o6[m+h]
+
303.0987;found 303.0982顺式-和反式氟醇a2的同步还原按照常规步骤c,二醇d2a和d2b分别环化为同一产品(35)。由d2b的sn2环化产生的α-异构体环化为热力学上更稳定的β-异构体35。按照常规步骤b,在-15℃下,将me4nhb(oac)3(0.174g,0.660mmol)和acoh(0.076ml,1.32mmol)加入到a2(0.040g,0.130mmol)在mecn(1.32ml)中的搅拌溶液中,并搅拌反应混合物24小时。通过闪蒸色谱法(戊烷:乙酸乙酯-1:3)纯化粗制的二醇d2a和d2b,得到白色固体二醇d2a和d2b(0.020克,50%,d.r.(顺/反)=1.2:1)。顺式-二醇、顺式-氟醇d2a的数据:1h nmr(600mhz,meod):δ7.76(d,j=8.0,1h),6.46(dd,j=44.4,4.8hz,1h),5.73(d,j=8.0hz,1h),4.03(ddd,j=18.3,7.0,5.0hz,1h),3.82(dd,j=11.4,5.1hz,1h),3.71(m,2h),3.60(dd,j=11.4,8.1hz,1h),1.42(s,3h),1.28(s,3h);
13
c nmr(150mhz,meod):δ165.8,151.7,143.1(d,j=2.6hz),102.9,100.1,94.3(d,j=208.4hz),74.6(d,j=24.6hz),73.7(d,j=4.5hz),67.3,65.3,28.3,19.7.hrms(ei
+
)计算得c
12h18
fn2o6[m+h]
+
305.1143;found 305.1142顺式-二醇、反式-氟醇d2b的数据:1h nmr(600mhz,meod):δ7.90(d,j=8.1hz,1h),6.71(dd,j=44.2,6.1hz,1h),5.74(d,j=8.1hz,1h),4.32(m,1h),3.81(m,3h),3.60(m,1h),1.43(s,3h),1.32(s,3h);
13
c nmr(150mhz,meod):δ165.8,152.2,143.0,
103.2100.2,92.6(d,j=204.4),75.9(d,j=2.8hz),71.5(d,j=29.1hz),65.7,64.5(d,j=2.2hz),28.6,19.4.hrms(ei
+
)计算得c
12h18
fn2o6[m+h]
+
305.1143;found 305.1123二醇d2a和d2b的环化按照常规步骤c,将d2(0.022克,0.072毫摩尔,d.r.顺/反=1.2:1)和2m naoh(0.36毫升,0.72毫摩尔)的溶液在mecn(0.72毫升)中搅拌24小时。通过闪蒸色谱法(ch2cl2:meoh-92.5:7.5)纯化粗产物35,得到白色固体核苷类似物35(0.019克,95%产率)。核苷类似物35的数据:[α]
d20
=+48.1(c 0.90in meoh);ir(neat):υ=2912,1436,1407,1042,952,697cm-1
;1h nmr(600mhz,(cd3)2co):δ7.71(d,j=8.0hz,1h),5.81(s,1h),5.61(d,j=8.0hz,1h),4.45(d,j=4.6hz,1h),4.20(dd,j=9.8,4.7hz,1h),4.12(dd,j=10.0,10.0hz,1h),3.90(dd,j=10.0,4.8hz,1h),3.86(ddd,j=10.0,10.0,4.7hz,1h),1.56(s,3h),1.42(s,3h);
13
c nmr(150mhz,(cd3)2co):δ164.2,151.8,142.4,103.4,102.3,94.5,75.3,74.6,72.5,66.1,33.1,22.8hrms(ei
+
)计算得c
12h17
n2o6[m+h]
+
285.1081;found 285.1085核苷类似物35的脱保护将35(0.019克,0.068毫摩尔)溶于meod(0.68毫升)中并加入两滴1m hcl,并将溶液在室温下放置12小时。随后,反应混合物在减压下浓缩,得到白色固体的核苷24(0.017克,100%)。所述光谱数据与以前的报告(46)相符。核苷24的数据:[α]
d20
=-23(c=0.1,meoh);ir(neat):ν=3347,2927,2857,1679,1464,1381,1260,1202,1104,1053,806cm
–1;1h nmr(600mhz,meod):δ8.03(d,j=8.1hz,1h),5.91(d,j=4.7hz,1h),5.70(d,j=8.1hz,1h),4.18(dd,j=4.9,4.9hz,1h),4.15(dd,j=4.9,4.9hz,1h),4.00-4.01(m,1h),3.84(dd,j=12.2,2.6hz,1h),3.74(dd,j=12.2,3.1hz,1h);
13
c nmr(150mhz,meod):166.2,152.5,142.7,102.6,90.6,86.4,75.7,71.3,62.3hrms(ei
+
)计算得c9h
13
n2o6[m+h]
+
245.0768;found 245.0770二醇d2a和d2b的相对立体化学的测定
基于化合物d5a/d5b、d8a/d8b的j-基构型分析和化合物18a、d7b、d9a的xrd分析,在氟甲基(fluoromethine)中心的立体化学和氟甲基质子的化学位移(*)之间建立了明确的趋势。在每一种情况下,顺式-氟醇二醇的化学位移都比非对映的反式氟醇二醇低。这里,d2a的化学位移为6.46ppm,而d2b的氟甲基质子的化学位移为6.71ppm。d2a被指定为顺式-氟醇二醇,d2b为反式-氟醇二醇。核苷35的相对立体化学的测定对核苷35的2dnoesy分析显示了所示的立体化学。此外,核苷24的1h nmr和
13
c nmr与报道(38)的数据相符。核苷对映-35的对映体过量测定按照常规步骤a、b和c,使用1:1的l-:d-脯氨酸,制备核苷对映-35的外消旋品。使用3μm amylose-1柱通过手性hplc分离核苷对映体;流速0.25ml/min;洗脱剂:己烷-iproh 85:15;检测波长254nm;保留时间:(-)-35为19.99min;(+)-35为23.30min。使用相同方法(95:5e.r.)测定光学富集的对映-35的对映体比率。羟醛缩合加成物a3、二醇加成物d3和核苷类似物na3和25的制备α-氟化/羟醛缩合按照文献步骤(47)制备相应的起始醛/水合物sm3。按照常规步骤a,将sm3(0.40毫摩尔)、nfsi(0.126克,0.40毫摩尔)、l-脯氨酸(0.046克,0.40毫摩尔)和nahco3(0.034克,0.40毫摩尔)的溶液在dmf(0.53毫升)中于4℃搅拌14小时。然后加入二氧环己酮(dioxanone)8(0.032毫升,0.27毫摩尔)在ch2cl2(0.67毫升)中的溶液,并将反应混合物在4℃下搅拌96小时。通过闪蒸色谱法(戊烷:乙酸乙酯-3:7)纯化粗制的氟醇a3(0.072克,产率84%,d.r.1.3:1)得到淡白色固体氟醇a3。2个非对映体和它们相应的同分异构体的混合物(1:1.1:0.65:0.28)。改变溶液的ph值会改变这些产物的比例。还原后,粗品中只有2个产品(d.r.(syn/anti)=1.3:1)存在。
顺式-和反式-氟醇a3的数据:ir(neat):υ=2995,1696,1451,1376,1087,1049cm-1
;1h nmr(600mhz,cdcl3):δ8.65,8.60,8.52,7.57,7.46,7.41,7.23,6.67,6.66,6.64,6.52,4.59,4.54,4.52,4.40,4.39,4.36,4.35,4.35,4.33,4.33,4.32,4.32,4.12,4.11,4.07,4.06,3.67,3.37,1.97,1.95,1.95,1.94,1.52,1.51,1.51,1.49,1.47,1.46,1.45,1.44;
13
c nmr(150mhz,cdcl3):δ211.4208.5,207.9,206.4,163.4,163.2,163.2,163.1,150.8,150.5,149.9,149.9,137.2,136.2,135.7,134.6,112.6,112.0,111.9,111.0,102.1,102.1,101.8,101.7,91.9,90.8,90.7,90.1,73.7,73.0,71.5,70.8,70.6,70.5,68.2,68.0,67.1,66.8,66.6,66.5,24.0,23.9,23.7,23.7,23.7,23.6,23.6,23.4,12.7,12.7,12.7,12.7;
19
f nmr(470mhz,cdcl3):δ
–
159.9,
–
161.6,
–
169.6,
–
177.8hrms(ei
+
)计算得c
13h18
fn2o6[m+h]
+
317.1143;found 317.1142顺式-氟醇和反式-氟醇a3的同步还原按照常规步骤b,在-15℃下,将me4nhb(oac)3(0.416克,1.58毫摩尔)和acoh(0.181毫升,3.16毫摩尔)加入到a3(0.100克,0.316毫摩尔)在mecn(2.10毫升)中的搅拌溶液中,然后搅拌反应混合物18小时。通过闪蒸色谱法(戊烷:乙酸乙酯-3:7)纯化粗制的二醇d3a得到白色固体d3a和d3b(0.063克,产率63%,d.r.(顺:反)=1.3:1)。顺式-二醇、顺式-氟醇d3a的数据:[α]
d20
=-11.8(c 1.0in meoh);ir(neat):υ=3363,2924,2858,1674,1380,1209,1075cm-1
;1h nmr(600mhz,cd3cn):δ7.42(d,j=0.90hz,1h),6.36(dd,j=44.9,5.1hz,1h),4.04(ddd,j=18.1,6.6,5.1hz,1h),3.79(dd,j=11.3,4.5hz,1h),3.67(m,2h),3.55(m,1h),1.83(d,j=0.90hz,3h),1.39(s,3h),1.24(s,3h);
13
c nmr(150mhz,cd3cn):δ164.7,151.5,137.9,111.7,99.9,94.0(d,j=205.9hz),74.8(d,j=25.1hz),73.0(d,j=4.3hz),67.1,65.0,28.8,19.9,12.7;
19
f nmr(470mhz,cd3cn):δ
–
169.1顺式-二醇,顺式-氟醇d3a在meod中的1h nmr用于相对立体化学的分配:1h nmr(600mhz,meod):δ7.58(s,1h),6.43(dd,j=4.1hz,1h),4.06(m,1h),3.81(m 1h),3.71(m,2h),3.59(m,1h),1.89(s,3h),1.41(s,3h),1.26(s,3h)。hrms(ei+)计算结果为c13h20fn2o6[m+h]+319.1300;found319.1329顺式-二醇、反式-氟醇d3b的数据:[α]
d20
=+26.2(c 0.45in ch3cn);ir(neat):υ=3360,2922,2855,1670,1380,1207,1078cm-1
;1h nmr(600mhz,meod):7.72(d,j=1.1hz,1h),6.71(dd,j=44.3,6.8hz,1h),4.32(m,1h),3.82(m,3h),3.60(m,1h),1.90(d,j=1.1hz,3h),1.44(s,3h),1.32(s,3h);
13
c nmr(150mhz,meod):δ166.1,152.5,138.3,112.0,
100.2,92.6(d,j=204.7hz),75.9,71.3(d,j=29.9hz),65.7,64.4(d,j=2.1hz),28.6,19.5,12.4.
19
f nmr(470mhz,cd3cn):δ
–
160.3.hrms(ei
+
)计算得c
13h20
fn2o6[m+h]
+
319.1300;found 319.1320二醇d3a和d3b的环化按照常规步骤c,二醇d3a和d3b分别环化成相同的产品na3。从d3b的sn2环化产生的α-异构体在环化后生成热力学上更稳定的β-异构体na3。按照常规步骤c,将d3a和d3b(0.100克,0.314毫摩尔,d.r.syn/anti=1.5:1)和2m naoh(0.236毫升,0.472毫摩尔)的溶液在mecn(3.14毫升)中搅拌10小时。通过闪蒸色谱法(乙酸乙酯)纯化粗制的核苷na3,得到白色固体核苷na3(0.089克,产率95%)。核苷na3的数据:[α]
d20
=+39.4(c 1.1in mecn);ir(neat):ν=3405,2993,1687,1267,1138,845,734cm
–1;1h nmr(600mhz,cd3cn):δ9.04(br s,1h),7.19(d,j=1.1hz,1h),5.67(s,1h),4.22(dd,j=4.8,3.1hz,1h),4.15(dd,j=9.1,3.5hz,1h),4.02(dd,j=10.1,9.8hz,1h),3.70(m,2h),3.55(m,1h),1.85(d,j=1.1hz,3h),1.53(s,3h),1.41(s,3h);
13
c nmr(150mhz,cd3cn):δ164.9,151.6,137.5,111.8,102.3,93.8,74.7,74.1,72.1,65.6,29.6,20.5,12.7hrms(ei
+
)计算得c
13h19
n2o6[m+h]
+
299.1238;found:299.1277.核苷类似物na3的脱保护将na3(0.010克,0.034毫摩尔)溶解于meod(0.34毫升)中并加入两滴1mhcl,然后将溶液在室温下放置12小时。随后,所述反应混合物在减压下浓缩,得到白色固体25(8.7毫克,100%)。光谱数据与以前的报告(48)相符。
核苷类似物25的数据:[α]
d20
=-33.0(c=0.1in meoh);ir(neat):ν=3346,2928,2867,1688,1466,1378,1262,1200,1104,1050,803cm
–1;1h nmr(600mhz,meod):δ7.86(d,j=1.1hz,1h),5.91(d,j=4.6hz,1h),4.15-4.18(m,2h),3.98-4.00(m,1h),3.86(dd,j=12.2,2.7hz,1h),3.75(dd,j=12.2,3.0hz,1h),1.88(d,j=0.9hz,3h);
13
c nmr(150mhz,meod):δ166.4,152.7,138.4,111.5,90.3,86.3,75.5,71.3,62.3,12.4.hrms(ei
+
)calcd for c
10h15
n2o6[m+h]
+
259.0925;found:259.0923.二醇d3a和d3b的相对立体化学的测定基于对化合物d5a/d5b、d8a/d8b的j-基构型分析和化合物18a、d7b、d9a的xrd分析,在氟甲基中心的立体化学和氟甲基质子的化学位移(*)之间建立了明确的趋势。在每一种情况下,顺式-氟醇二醇的化学位移都比非对映的反式-氟醇二醇低。这里,d3a的化学位移为6.43ppm,而d3b的氟甲基质子的化学位移为6.69ppm。d3a被指定为顺式-氟醇二醇,d3b为反式-氟醇二醇。绝对立体化学的测定将核苷25的[α]
d20
值与文献值进行比较,证实了绝对立体化学(49)。核苷na3对映体过量的测定按照常规步骤a、b和c,使用l-:d-脯氨酸的1:1混合物,制备核苷na3的外消旋品。使用3μm直链淀粉-1柱通过手性hplc分离对映体核苷;流速0.25ml/min;洗脱剂:己烷-iproh85:15;检测波长254nm;保留时间:(+)-na3为5.18min;(-)-na3为12.61min。采用相同的方法(91:9e.r.)测定光学富集的(+)-na3的对映体比例。羟醛缩合加合物a4、二醇加合物d4a/d4b和核苷类似物27的制备顺式-和反式氟醇的α-氟化/羟醇缩合和同步还原按照常规步骤a,2-(4,6-二氯嘧啶-5-基)乙醛(2-(4,6-dichloropyrimidin-5-yl)acetaldehyde)(0.250g,1.31mmol,1当量),nfsi(0.413g,1.31mmol,1当量),l-脯氨酸(0.151g,1.31mmol,1当量)和碳酸氢钠(0.110g,1.31mmol,1当量)的溶液在dmf(1.19ml)中于4℃搅拌1小时。将二氧环己酮8(0.521ml,4.36mmol,3.33当量)加入反应混合物中,在4℃下搅拌24小时。用闪蒸色谱法(戊烷:乙酸乙酯-3:7)纯化粗氟醇a4,得到橙油状的氟醇a4(0.301g,产率68%)。按照常规步骤b,在-15℃下,将me4nhb(oac)3(2.16g,8.21mmol)和acoh(0.905ml,16.4mmol)加入a4(0.555g,1.64mmol)在mecn(16.4ml)中的搅拌溶液中,并将反应混合物搅拌24小时。用闪蒸色谱法(戊烷:乙酸乙酯-4:1)纯化粗二醇d4a,得到灰白色固
体二醇d4a(0.295g,产率53%,d.r,(syn/anti)=3:1)。顺式-二醇d4a的数据:[α]
d20
=+26.6(c 5.0in mecn);ir(neat):υ=3000,1442,1375,1039,918,cm-1
;1h nmr(600mhz,cdcl3):δ8.73(s,1h),6.05(dd,j=46.0,7.9hz,1h),4.64(m,1h),3.89(dd,j=11.5,5.7hz,1h),3.80(m,1h),3.73(dd,j=9.1,8.5hz,1h),3.61(dd,j=11.5,9.5hz,1h),1.29(s,3h),0.94(s,3h);
13
c nmr(150mhz,cdcl3):δ161.5,157.4,127.8,98.3,91.1(d,j=179.4hz),75.5(d,j=21.3hz),71.7(d,j=5.5hz),66.6,63.3,28.2,18.7;
19
f nmr(470mhz,cdcl3):δ
–
193.0.hrms(ei
+
)calcd for c
12h16cl2
fn2o4[m+h]
+
341.0466;found 341.0425二醇d4a的环化按照常规步骤c,d4a(0.014g,0.044mmol,1当量)和2m氢氧化钠(0.11ml,0.22mmol,5当量)的溶液在mecn(0.30ml)中搅拌15分钟。用闪蒸色谱法(乙酸乙酯:戊烷-50:50)纯化粗核苷27得到白色固体核苷27(6.4mg,51%的产率)。核苷类似物27的数据:[α]
d20
=+51.2(c 0.34in ch2cl2);ir(neat):υ=3363,2927,1602,1598,1571,1408,968cm-1
;1h nmr(600mhz,cd3cn):δ8.66(s,1h),4.19(dd,j=10.1,4.9hz,1h),3.91(dd,j=10.2,10.1hz,1h),3.86(dd,j=10.1,4.7hz,1h),3.30(ddd,j=10.2,10.1,4.8hz,1h),1.56(s,3h),1.51(s,3h);
13
c nmr(150mhz,cd3cn):δ176.6,160.8,158.7,114.6,101.7,82.2,79.1,75.6,69.0,64.7,28.9,19.5.hrms(ei
+
)calcd for c
12h14
cln2o4[m+h]
+
285.0637;found 285.0644核苷27的相对立体化学的测定核苷27的2d noesy分析显示了所示的立体化学。二醇d4a对映体过量的测定按照常规步骤a和b,使用l-:d-脯氨酸的1:1的混合物,制备二醇d4a的外消旋品。使用3μm直链淀粉-1柱通过手性hplc分离对映体二醇;流速0.25ml/min;洗脱剂:己烷-iproh90:10;检测波长254nm;保留时间:(-)-d4a为11.81min;(+)-d4a为12.68min。采用相同的方法(95:5e.r.)测定光学富集物的(+)-d4a的对映体比例。
s5、水合物sm5、羟醛缩合加合物a5、二醇加合物d5a和d5b以及核苷类似物28的制备1,2,3-三唑(1.00ml,17.2mmol,1.0当量),溴乙醛二乙基缩醛(3.10ml,20.7mmol,1.2当量)和碳酸钾(4.75g,34.4mmol,2.0当量)的溶液在dmf(86ml)中于90℃搅拌24小时。然后用40ml的二氯甲烷过滤和洗涤反应混合物,并进行减压浓缩。用闪蒸色谱法纯化粗产物s5(戊烷:乙酸乙酯-7:3)得到无色油状的s5(2.90克,产量91%)。s5(0.100g,0.54mmol,1.0当量)在0.5m hcl(0.54ml)中的溶液加热至90℃并持续5小时。当反应混合物完全转化为sm5后,减压浓缩并将所得产物sm5用于反应,无需提纯。s5的数据:1h nmr(400mhz,cdcl3):δ7.68(d,j=0.90hz,1h),7.66(d,j=0.90hz,1h),4.76(t,j=5.3hz,1h),4.48(d,j=5.3hz,2h),3.73(m,2h),3.47(m,2h),1.17(m,6h);
13
c nmr(125mhz,cdcl3):δ133.8,124.9,101.1,64.0,52.9,15.3.hrms(ei
+
)calcd for c8h
16
n3o2[m+h]
+
186.1237;found 186.1233α-氟化/羟醛缩合按照常规步骤a,在4℃下,将s5(0.54mmol)、选择氟(selectfluor)(0.192g,0.54mmol)、l-脯氨酸(0.063g,0.54mmol)和碳酸氢钠(0.045g,0.54mmol)的溶液在dmf(0.72ml)中搅拌12小时。然后加入二氧环己酮8(0.043ml,0.36mmol)的mecn(0.43ml),然后在室温下搅拌反应混合物72小时。用闪蒸色谱法(et2o)纯化粗氟醇a5得到浅黄色的油状氟醇a5(0.061g,产率65%,d.r,1:1)。顺式-和反式-氟醇a5的数据:ir(neat):υ=3138,2990,1749,1455,1379,1224,1070,799cm-1
;1h nmr(600mhz,cdcl3):δ8.24(1h),8.12(1h),7.79(1h),7.77(1h),6.89(1h),6.86(1h),4.74(1h),4.49(1h),4.33(2h),4.26(1h),4.14(1h),4.06(1h),3.89(1h),1.55(3h),1.48(3h),1.44(3h),1.31(3h);
13
c nmr(150mhz,cdcl3):δ210.8,209.4,134.5,134.5,124.4,124.4,102.1,102.0,94.5,93.5,72.1,71.3,70.8,70.1,66.5,66.5,23.8,23.5,23.4,23.4;
19
f nmr(470mhz,cdcl3):δ-154.6,-163.8.hrms(ei
+
)calcd for c
10h15
fn3o4[m+h]
+
260.1041;found 260.1044顺式-和反式-氟醇a的同步还原按照常规步骤b,在-15℃下,将me4nhb(oac)3(0.391g,1.49mmol)和acoh(0.170ml,2.98mmol)加入到a5(0.077g,0.30mmol)在mecn(3.00ml)中的搅拌溶液中,得到的混合物被搅拌24小时。用闪蒸色谱法(ch2cl2:meoh-96:4)纯化粗二醇d5a和d5b,得到白色固体二醇d5a和d5b(0.072g,产率94%,d.r.(syn/anti)=1.2:1)。
顺式-二醇、顺式-氟醇d5a的数据:[α]
d20
=+52.4(c 0.51in mecn);ir(neat):υ=3432,2997,2253,1444,1375,1071,1039cm-1
;1h nmr(600mhz,cd3cn):δ8.17(d,j=1.0hz,1h),7.78(d,j=1.0hz,1h),6.69(dd,j=48.1,4.7hz,1h),4.36(ddd,j=18.4,5.0,5.0hz,1h),3.79(dd,j=11.4,5.0hz,1h),3.63(m,2h),3.54(m,2h),1.39(s,3h),1.31(s,3h);
13
c nmr(150mhz,cd3cn):δ135.2,126.2,100.0,95.9(d,j=206.7hz),74.7(d,j=22.7hz),73.1(d,j=4.4hz),66.0,65.2,28.8,19.9;
19
f nmr(470mhz,cdcl3):δ-156.0hrms(ei
+
)calcd for c
10h17
fn3o4[m+h]
+
262.1198;found 262.1209.顺式-二醇、反式-氟醇d5b的数据:[α]
d20
=+40.0(c 0.37in mecn);ir(neat):υ=3000,1442,1375,1039,918,740cm-1
;1hnmr(600mhz,cd3cn):δ8.22(d,j=1.0hz,1h),7.79(d,j=1.0hz,1h),6.78(dd,j=46.4,6.0hz,1h),4.53(ddd,j=10.4,6.0,4.7hz,1h),4.09(br s,1h),3.83(m,2h),3.57(m,2h),3.41(br s,1h),1.35(s,3h),1.34(s,3h);
13
c nmr(150mhz,cd3cn):δ135.3,125.7,100.0,96.5(d,j=204.3hz),74.2(d,j=2.3hz),72.9(d,j=27.2hz),65.4,65.3(d,j=2.0hz),28.9,19.8;
19
f nmr(470mhz,cdcl3):δ-151.2hrms(ei
+
)calcd for c
10h17
fn3o4[m+h]
+
262.1198;found 262.1206二醇d5a的环化按照常规步骤d,二醇d5a被单独环化为28,而二醇d5b没有环化。这表明,由二醇混合物生成的产物仅由d5a二醇经sn2环化而成。按照常规步骤d,将d5a和d5b(0.025g,0.096mmol,1.0当量,d.r.(syn/anti)=
1.2:1)和sc(otf)3(0.118g,0.239mmol,2.5当量)的溶液在干燥mecn(1.00ml)中搅拌。12小时后,加入吡啶(0.50ml)和乙酸酐(0.25ml),并将反应混合物搅拌3小时。用闪蒸色谱法(戊烷:乙酸乙酯-1:3)纯化粗28,得到透明无色油状的核苷类似物28(0.015g,产率47%)。核苷类似物28的数据:[α]
d20
=+1.3(c 0.60in ch2cl2);ir(neat):υ=2926,1747,1373,1227,1064cm-1
;1h nmr(600mhz,cdcl3):δ7.76(s,1h),7.26(s,1h),6.19(d,j=3.7hz.1h),5.85(dd,j=5.0,3.8hz,1h),5.63(dd,j=5.3,5.0hz,1h),4.49(ddd,j=5.3,4.3,3.0hz,1h),4.41(dd,j=12.4,3.0hz,1h),4.22(dd,j=12.4,4.3hz,1h),2.13(s,3h),2.13(s,3h),2.06(s,3h);
13
c nmr(150mhz,cdcl3):δ170.5,169.6,169.5,134.3,122.9,90.0,81.0,74.5,70.8,62.9,20.8,20.6,20.6;hrms(ei
+
)calcd for c
13h18
n3o7[m+h]
+
328.3005;found 328.3000二醇d5a的相对立体化学的测定采用j-基构型分析确定了二醇d5a的相对立体化学。有关详细信息,请参见基于j的配置分析部分。二醇d5b的相对立体化学性质的测定采用j-基构型分析确定二醇d5b的相对立体化学。有关详细信息,请参见基于j的配置分析部分。核苷28的相对立体化学的测定核苷28a的2d noesy分析支持了所示的立体化学。二醇d5a对映体过量的测定
按照常规步骤a和b,使用1:1的l-:d-脯氨酸的混合物,制备二醇d5a的外消旋品。使用3μmi-cellulose-5柱通过手性hplc分离对映体二醇;流速0.20ml/min;洗脱剂:己烷-iproh 90:10;检测波长210nm;保留时间(+)-d5a为4.69min;(-)-d5a的保留时间为5.80min。使用相同的方法(93:7e.r.)测定光学富集的(+)-d 5a二醇的对映体比例。二醇d5b对映体过量的测定按照常规步骤a和b,使用1:1的l-:d-脯氨酸的混合物,制备二醇d5b的外消旋品。使用3μmi-cellulose-5柱通过手性hplc分离对映体二醇;流速0.20ml/min;洗脱剂:己烷-iproh 90:10;检测波长210nm;保留时间:(-)-d5b为3.94min;(+)-d5b的保留时间为4.95min。使用相同的方法(96:4e.r.)测定光学富集的(+)-d5b二醇的对映体比例。ent-d5a二醇对映体过量的测定按照常规步骤a和b,使用1:1的l-:d-脯氨酸的混合物,制备二醇ent-d5a的外消旋品。使用3μm i-cellulose-5色谱柱通过手性hplc分离对映体二醇;流速0.20ml/min;洗脱剂:己烷-iproh 90:10;检测波长210nm;保留时间:(+)-d5a为4.69min;(-)-d5a为5.80min。采用相同的方法(95:5e.r.)测定光学富集的ent-d5a二醇的对映体比例。ent-d5b二醇对映体过量的测定按照常规步骤a和b,使用1:1的l-:d-脯氨酸的混合物,制备二醇ent-d5b的外消旋品。使用3μm i-cellulose-5柱通过手性hplc分离对映体二醇;流速0.20ml/min;洗脱剂:己烷-iproh 90:10;检测波长210nm;保留时间:(-)-d5b为3.94min;(+)-d5b的保留时间为4.95min。采用相同的方法(95:5e.r.)测定光学富集的e nt-d5b二醇的对映体比率。s6、水合物sm6、羟醛缩合加合物a6、二醇加合物d6a和d6b,以及核苷类似物29的制备三氟甲基尿嘧啶(trifluoromethyluracil)(1.00g,5.52mmol,1.0当量),溴乙醛二乙基缩醛(1.66ml,11.1mmol,2.0当量)和碳酸钾(1.53g,11.1mmol,2.0当量)的溶液在dmf(27.6ml)中于90℃搅拌24小时。然后用40ml的ch2cl2过滤和洗涤反应混合物,并进行减压浓缩。用闪蒸色谱法(戊烷:乙酸乙酯-7:3)纯化粗s6,得到无色油状的s6(0.605g,产率37%)。s7溶液(0.100g,0.340mmol,1.0当量)在0.5m hcl(0.34ml)中被加热到90℃并持续5小时。在完全转化为醛/水合物sm6后,将反应混合物在减压下浓缩,所得到的醛/水合物sm6可直接用于反应,无需纯化。s6的数据:ir:υ=3430,2988,2800,1109,1025cm-1
;1h nmr(600mhz,cdcl3):δ8.56(brs,1h),7.82(s,1h),4.61(t,j=5.0hz),3.88(d,j=5.0hz),3.78(m,2h),3.54(m,2h),1.21(m,6h);
13
c nmr(150mhz,cdcl3):δ158.6,150.0,147.0(q,j=5.8hz),121.9(q,j=270.5hz),104.7(q,j=33.5hz),100.0,64.6,51.0,15.3.hrms(ei
+
)calcd for c
11h16
f3n2o4[m+h]
+
297.1057;found 297.1056α-氟化/羟醛缩合
按照常规步骤a,在4℃,将sm6(0.340mmol)、nfsi(0.107g,0.340mmol)、l-脯氨酸(0.039g,0.340mmol)和碳酸氢钠(0.029g,0.340mmol)的溶液在dmf(0.45ml)中搅拌12小时。然后加入二氧环己酮8(0.027ml,0.227mmol)的二氯甲烷(0.57ml)溶液,将反应混合物在4℃下搅拌96小时。用闪蒸色谱法纯化粗氟醇a6(戊烷:乙酸乙酯-65:35),得到浅黄色油状的氟醇a6(0.050g,产量60%)。顺式-和反式-氟醇a6的数据:ir:υ=2991,1699,1450,1087,1049cm-1
;1h nmr(600mhz,cd3cn):δ9.53,9.52,8.15,8.11,6.58,6.46,4.62,4.56,4.55,4.43,4.31,4.29,3.98,3.98,1.43,1.40,1.40,1.38;
13
c nmr(150mhz,cd3cn):δ208.4,,207.9,159.6,159.5,150.6,150.1,144.0,144.0,123.6,123.5,106.6,106.0,102.4,102.3,95.3,92.4,76.3,76.1,69.9,69.1,67.9,67.8,24.5,24.4,24.2,23.9;
19
f nmr(470mhz,cd3cn):δ
–
64.1,
–
64.1,
–
161.4,
–
169.1.hrms(ei
+
)calcd for c
13h14
f4n2nao6[m+na]
+
393.0680;found 393.0682顺式-和反式-氟醇a6的同步还原按照常规步骤b,在-15℃下,将me4nhb(oac)3(0.355g,1.35mmol)和acoh(0.155ml,2.79mmol)加入到a6(0.100g,0.27mmol,1当量)在mecn(1.80ml)中的搅拌溶液中。然后将反应混合物搅拌24小时。用闪蒸色谱法(戊烷:乙酸乙酯-4:1)对粗二醇d6a和d6b进行纯化,得到白色固体二醇d6a(0.040g,产率40%)和d6b(0.019g,产率19%)。顺式-二醇、顺式-氟醇d6a的数据:[α]
d20
=+18.4(c 0.50in ch2cl2);ir(neat):υ=3426,2996,1702,1463,1379,1070cm-1
;1h nmr(600mhz,cd3cn):δ9.42(br s,1h),8.10(s,1h),6.33(dd,j=45.1,5.6hz,1h),4.28(dd,j=14.8,5.6hz,1h),3.79(dd,j=11.15.5hz,1h),3.70(m,2h),3.60(dd,j=9.5,2.7hz,1h),3.55(dd,j=10.4,9.5hz,1h),1.35(s,3h),1.30(s,3h);
13
c nmr(150mhz,cd3cn):δ159.5,150.1,144.2(q,j=6.3hz),123.5(q,j=266.4hz),106.3(q,j=32.9hz),99.9,96.3(d,j=210.9hz),73.9(d,j=3.8hz),70.5(d,j=24.5hz),65.4,63.0,29.1,19.8;
19
f nmr(470mhz,cd3cn):δ
–
64.1,
–
168.0.hrms(ei
+
)calcd for c
13h17
f4n2nao6[m+na]
+
395.0837;found 395.0836.
顺式-二醇、反式-氟醇d6b的数据:[α]
d20
=-37.2(c 1.1in ch2cl2);ir(neat):υ=3424,1703,1466,1379,1281,1138,1042cm-1
;1h nmr(600mhz,cd3cn):δ8.26(s,1h),6.67(dd,j=43.0,4.9hz,1h),4.34(m,1h),3.78(dd,j=11.2,5.1hz,1h),3.72(m,2h),3.54(dd,j=11.2,8.3hz,1h),1.39(s,3h),1.26(s,3h);
13
c nmr(150mhz,cd3cn):δ159.5,150.6,144.2,123.6(q,j=272.9hz),105.9(q,j=32.5hz),100.0,92.5(d,j=206.1hz),74.2(d,j=4.4hz),72.3(d,j=27.7hz),65.4,64.8,29.0,19.7;
19
f nmr(470mhz,cd3cn):δ
–
64.1,
–
161.7.hrms(ei
+
)calcd for c
13h17
f4n2nao6,[m+na]
+
395.0837;found 395.0838.二醇d6a和d6b的环化按照常规步骤d,二醇d6b被单独环化为29,而二醇d6a没有环化。这表明,由二醇混合物产生的产物仅由d6b二醇经sn2环化而成。按照常规步骤d,将d6a和d6b(0.045g,0.121mmol,d.r.(syn/anti)=1:2)和sc(otf)3(8.9mg,0.018mmol,0.15当量)的溶液在干燥的mecn(1.21ml)中搅拌24小时。通过闪蒸色谱法(戊烷:乙酸乙酯-3:7)纯化粗29,得到无色油状的核苷29(0.013g,产量45%(来自反式-氟醇d6b))。核苷类似物29的数据:[α]
d20
=-16.7(c 0.49in ch2cl2);ir(neat):υ=3405,2924,2854,1702,1465,1276cm-1
;1hnmr(600mhz,cd3cn):δ9.33(br s,1h),7.97(q,j=1.2hz,1h),6.18(d,j=4.1hz,1h),4.86(m,2h),4.42(dd,j=3.6,2.4hz,1h),3.67(m,2h),
3.21(dd,j=5.6,4.4hz,1h),1.36(s,3h),1.30(s,3h);
13
c nmr(150mhz,cd3cn):δ159.4,149.9,143.6(q,j=6.0hz),123.6(q,j=269.7hz),113.6,103.4(q,j=33.2hz),87.7,84.7,82.8,80.2,64.0,25.7,24.0;
19
f nmr(470mhz,cd3cn):δ
–
63.8hrms(ei
+
)calcd for c
13h16
f3n2o6[m+h]
+
353.0955;found 353.0971核苷29的相对立体化学的测定核苷29的2d noesy分析支持了所示的立体化学。二醇d6a和d6b的相对立体化学的测定基于对化合物d5a/d5b、d8a/d8b的j-基构型分析和化合物18a、d7b、d9a的xrd分析,在氟甲基中心的立体化学和氟甲基质子的化学位移(*)之间建立了明确的趋势。在每一种情况下,顺式-氟醇二醇的化学位移比反式-氟醇二醇的低。在这里,d6a的化学位移为6.33ppm,而d6b的氟甲基质子的化学位移为6.67ppm。d6a为顺式-氟醇二醇,d6b为反式-氟醇二醇。核苷29对映体过量的测定按照常规步骤a、b和c,使用1:1的l-:d-脯氨酸混合物,制备核苷29的外消旋品。使用3μm直链淀粉-1柱通过手性hplc分离对映体核苷;流速0.25ml/min;洗脱剂:己烷-iproh90:10;检测波长254nm;保留时间:(+)-29为9.10min;(-)-29保留时间为13.14min。采用相同方法(94:6e.r.)测定光学富集的(-)-29核苷的对映体比例。s7、水合物sm7、羟醛缩合加合物a7、二醇加合物d7a和d7b以及核苷类似物30的制备。顺式-和反式-氟醇a的α-氟化/羟醛缩合和同步还原按照常规步骤a,邻苯二甲胺乙醛(phthalimidoacetaldehyde)(0.100g,0.529mmol,1.5当量),nfsi(0.167g,0.529mmol,1.5当量),l-脯氨酸(0.061g,0.529mmol,1.5当量)和2,6-二甲基吡啶(0.061ml,0.529mmol,1.5当量)的溶液在dmf(0.71ml)中于4℃搅拌12小时。加入二氧环己酮8(0.042ml,0.353mmol,1当量)的二氯甲烷(0.88ml)溶液,在室温下搅拌反应混合物48小时。用闪蒸色谱法(戊烷:乙酸乙酯-1:1)纯化粗氟醇a7,得到黄色油状的氟醇a7(0.069g,产率58%,d.r.2.2:1)。按照常规步骤b,在-15℃下,将me4nhb(oac)3(0.776g,2.95mmol)和acoh(0.337ml,5.90mmol)加入到a7(0.200g,0.59mmol)在mecn(5.90ml)中的搅拌溶液中,并将反应混合物搅拌24小时。用闪蒸色谱法(戊烷:乙酸乙酯-3:7)纯化粗二醇d7a和d7b,得到白色固体二醇d7a和d7b(0.094g,产率47%,d.r.(syn/
anti)=1.5:1)。顺式-二醇、顺式-氟醇d7a的数据:[α]
d20
=-11.4(c 2.0in ch2cl2);ir(neat):υ=3442,2992,1785,1724,1377,1074,721cm-1
;1h nmr(600mhz,cd3cn):δ7.93(m,2h),7.89(m,2h),6.07(dd,j=48.6,7.9hz,1h),4.76(m,1h),4.43(m,1h),3.73(m,2h),3.58(dd,j=8.8,6.0hz,1h),3.47(m,1h),3.41(m,1h),1.21(s,3h),0.92(s,3h);
13
c nmr(150mhz,cd3cn):δ167.8(d,j=1.5hz),136.0,132.5,124.6,99.1,91.1(d,j=202.0hz),73.3(d,j=6.6hz),71.8(d,j=25.3hz),65.1,64.5,28.1,19.3;
19
f nmr(470mhz,cd3cn):δ
–
157.8hrms(ei
+
)calcd for c
16h19
fno6[m+h]
+
340.1191;found 340.1190.顺式-二醇、反式-氟醇d7b的数据:[α]
d20
=-1.0(c 2.3in ch2cl2);ir(neat):υ=3442,2992,1784,1725,1375,1070,723cm-1
;1h nmr(600mhz,cd3cn):δ7.94(m,2h),7.89(m,2h),6.34(dd,j=46.0,9.2hz,1h),4.80(m,1h),3.92(ddd,j=9.5,1.8,1.4hz,1h),3.84(m,2h),3.73(m,1h),3.60(dd,j=10.8,8.7hz,1h),3.30(m,1h),1.47(s,3h),1.35(s,3h);
13
c nmr(150mhz,cd3cn):δ168.1(d,j=1.6hz),136.0,132.3,124.6,99.4,89.5(d,j=202.4hz),75.1,68.7(d,j=31.7hz),65.3,63.1(d,j=3.1hz),28.6,19.5;
19
f nmr(470mhz,cdcl3):δ
–
159.8.hrms(ei
+
)calcd for c
16h19
fno6[m+h]
+
340.1191;found 340.1172二醇d7a和d7b的环化
按照常规步骤d,二醇d7a被环化为30,而二醇d7b环化为30及其相应的α-异聚体的混合物。所述二醇混合物即来自二醇经sn2环化又来自二醇α-异构体的一些异构化。已有关于核苷的热处理(emperization)的报道(31)。按照常规步骤d,将d7a和d7b(0.033g,0.097mmol,1.0当量,d.r.(syn/anti)=2:1)和sc(otf)3(0.120g,0.243mmol,2.5当量)的溶液在mecn(0.65ml)中搅拌。然后加入0.25ml吡啶和0.25ml乙酸酐并搅拌1.5小时。通过闪蒸色谱法(戊烷:乙酸乙酯
–
7:3)纯化粗产物30得到无色油状的核苷类似物30(0.027g,产率69%)。核苷类似物30的数据:[α]
d20
=-9.0(c 1.96in ch2cl2);ir(neat):υ=2922,1781,1744,1721,1374,1222,1047,720cm-1
;1h nmr(500mhz,cdcl3):δ7.88(m,2h),7.77(m,2h),5.94(dd,j=6.0,4.1hz,1h),5.87(d,j=4.1hz,1h),5.65(dd,j=6.1,6.0hz,1h),4.49(dd,j=12.1,3.4hz,1h),4.29(ddd,j=9.5,5.9,3.4hz,1h),4.21(dd,j=12.1,5.9,1h),2.12(s,3h),2.11(s,3h),2.09(s,3h);
13
c nmr(150mhz,cdcl3):δ170.9,169.8,169.7,166.9,134.8,131.7,124.0,82.8,79.2,72.0,70.6,63.2,20.9,20.7,20.7.hrms(ei
+
)calcd for c
19h23
n2o9[m+nh4]
+
423.1398;found 423.1378二醇d7b的相对立体化学的测定乙醇中的再结晶允许使用单x射线晶体学指定相对立体化学。核苷30的相对立体化学的测定核苷30的2d noesy分析支持了所示的立体化学。二醇ent-d7a对映体过量的测定按照常规步骤a和b,使用1:1的l-:d-脯氨酸的混合物,制备二醇d7a的外消旋品。使用3μm amylose-1柱通过手性hplc对对映体核苷进行分离;流速0.25ml/min;洗脱剂:己烷-iproh 90:10;检测波长254nm;保留时间:(-)-d7a的为9.10min;(+)-d7a为
13.14min。采用相同的方法(95:5e.r.)测定光学富集的(+)-d7a二醇的对映体比例。sm8、羟醛缩合加合物a8、二醇加合物d8a/d8b和核苷类似物32/33的制备脱氮腺嘌呤(0.500g,1.79mmol,1.0当量),溴乙醛二乙基缩醛(0.323ml,2.15mmol,1.25当量)和碳酸钾(0.491g,3.58mmol,2.0当量)的溶液在dmf(9.00ml)中于90℃搅拌24小时。然后用10ml的二氯甲烷过滤和洗涤反应混合物,并进行减压浓缩。用闪蒸色谱法(戊烷:乙酸乙酯-7:3)纯化粗产物s8得到白色固体s8(0.375g,产率53%)。然后s8溶液(17.0g,43.0mmol,1.0当量)在2.0m hcl(129ml,258mmol,6.0当量)中加热到70℃并持续1小时。然后将反应混合物冷却到室温,再继续搅拌2小时。所示反应混合物在-20℃下保存过夜,然后过滤形成的沉淀物,用1:1的二恶烷:水(10mlx2)洗涤。在减压下干燥滤液sm8,并在反应中使用所得产物sm8(7.88g,产率为54%),无需纯化。s8的数据:1h nmr(600mhz,cdcl3):δ8.61(s,1h),7.50(s,1h),4.67(t,j=5.1hz,1h),4.35(d,j=5.1hz,2h),3.73(m,2h),3.48(m,2h),1.16(m,6h);
13
c nmr(150mhz,cdcl3):δ152.7,151.1,150.8,136.3,116.9,100.7,63.9,50.6,47.7,15.3.hrms(ei
+
)calcd for c
12h16
clin3o2[m+h]
+
395.9970;found 395.9973α-氟化/羟醛缩合按照常规步骤a,sm8(2.00g,5.86mmol,1当量),nfsi(1.85g,5.86mmol,1.0当量),l-脯氨酸(0.674g,5.86mmol,1.0当量)和碳酸氢钠(0.984g,11.71mmol,2.0当量)的溶液在dmf(10ml)中于20℃中搅拌18小时。将二氧环己酮8(0.762g,5.86mmol,1.0当量)加入反应混合物中,在室温下搅拌36小时。用闪蒸色谱法(戊烷中25-75%的乙酸乙酯)纯化粗产物a8得到浅黄色固体顺式-和反式-氟醇a8(1.58g,产率57%,d.r 1.2:1)。顺式-和反式-氟醇a8的数据:ir(neat):υ=3145,2988,1747,1575,1539,1444,1205,1084,949,734cm-1
;1h nmr(600mhz,dmso-d6):δ8.76,8.74,8.39,8.24,6.89,6.85,6.37,6.12,4.98,4.76,4.61,4.32,4.30,4.05,3.95,3.93,1.40,1.34,1.33,1.31
13
c nmr(150mhz,dmso-d6):δ206.3,206.1,151.6,151.5,151.3,151.2,151.0,134.5,134.1,116.8,116.7,100.4,100.1,91.4,09.4,76.1,74.7,68.7,68.0,66.6,66.4,55.3,55.1,24.6,24.1,22.9,22.7
19
f nmr(470mhz,dmso-d6):δ
–
146.0,
–
152.6.hrms(ei
+
)calcd for c
14h15
clfin3o4[m+h]
+
469.9774;found 469.9779顺式-和反式-氟醇a8的同步还原按照常规步骤b,在0℃,将nahb(oac)3(0.316g,1.49mmol,5当量)和acoh
(0.171ml,2.98mmol,10当量)的溶液加入到a8(0.140g,0.298mmol,1当量)在mecn(2.8ml)中的搅拌溶液中。然后在室温下搅拌反应混合物2小时。用闪蒸色谱法(戊烷:乙酸乙酯-70:30)纯化粗二醇d8a和d8b,得到白色固体二醇d8a和d8b(0.141克,77%的产量,d.r.(syn/anti)=1.5:1)。顺式-二醇、顺式-氟醇d8a的数据:[α]
d20
=-19.6(c 2.0in ch2cl2);ir(neat):υ=3335,2989,2890,1577,1540,1445,1206,1076,951cm-1
;1h nmr(600mhz,dmso-d6):δ8.73(s,1h),8.27(s,1h),6.73(dd,j=49.4,7.0hz,1h),6.08(br s,1h),4.84(d,j=4.1hz,1h),4.59(m,1h),3.59(m,1h),3.44(m,1h),3.42(m,1h),3.33(m,1h),1.16(s,3h),1.13(s,3h);
13
c nmr(150mhz,dmso-d6):δ151.4,151.2,151.1,134.5,116.7,97.8,92.0(d,j=203.3),73.2(d,j=5.7hz),71.0(d,j=24.2hz),63.8,62.5,54.9,28.0,19.1;
19
f nmr(470mhz,dmso-d6):δ
–
147.1.hrms(ei
+
)calcd for c
14h15
clfin3o4[m+h]
+
471.9931;found 471.9940.顺式-二醇、反式-氟醇d8b的数据:[α]
d20
=-11.6(c 0.38in ch2cl2);ir(neat):υ=3363,2931,2890,1579,1540,1444,1212,1067,951cm-1
;1h nmr(600mhz,dmso-d6):δ8.73(s,1h),8.34(s,1h),6.97(dd,j=46.9,7.9hz,1h),5.74(d,j=5.7hz,1h),5.22(d,j=5.7hz,1h),4.61(m,1h),3.84(m,1h),3.72(m,1h),3.52(dd,j=11.7,8.7hz,1h),1.35(s,3h),1.20(s,3h);
13
c nmr(150mhz,dmso-d6):δ151.5,151.4,151.2,134.1,116.6,97.9,90.9(d,j=203.5hz),74.3,69.1(d,j=30.3hz),64.2,61.4,54.8,28.4,19.0;
19
f nmr(470mhz,,dmso-d6):δ
–
146.3.hrms(ei
+
)calcd for c
14h15
clfin3o4[m+h]
+
471.9931;found 471.9940二醇d8a的环化按照常规步骤d,二醇d8a环化为32,而二醇d8b环化为33。这支持sn2环化而无后续
差向异构化。按照常规步骤d,d8a(0.050g,0.106mmol,1.0当量)和incl3(2.3mg,0.011mmol,0.10当量)的溶液在干燥mecn(1.00ml)中搅拌16小时。通过闪蒸色谱法(戊烷中20-80%的乙酸乙酯)纯化粗核苷32得到白色固体核苷32(0.029g,61%的产率)。核苷类似物32的数据:[α]
d20
=-23.9(c 0.46in ch2cl2);ir(neat):υ=3339,3113,2935,1576,1539,1445,1207,1108,951cm-1
;1h nmr(600mhz,dmso-d6):δ8.69(s,1h),8.23(s,1h),6.34(d,j=3.1hz,1h),5.19(dd,j=6.3,3.1hz,1h),5.14(br s,1h),4.94(dd,j=6.3,2.9hz,1h),4.20(m,1h),3.56(m,2h),1.54(s,3h),1.31(s,3h);
13
c nmr(150mhz,dmso-d6):δ151.2,150.8,150.4,133.9,116.7,113.2,89.4,86.3,83.9,80.9,61.4,53.7,27.0,25.1.hrms(ei
+
)calcd for c
14h16
clin3o4[m+h]
+
451.9869;found 451.9875二醇d8b的环化按照常规步骤d,d8b(0.050g,0.106mmol,1.0当量)和incl3(2.3mg,0.011mmol,0.10当量)的溶液在干mecn(1.00ml)中搅拌16小时。通过闪蒸色谱法(戊烷中20-80%的乙酸乙酯)纯化粗核苷33得到白色固体核苷33(0.034g,70%产率)。核苷类似物33的数据:[α]
d20
=-47.8(c 0.51in chcl3);1h nmr(600mhz,dmso-d6):δ8.66(s,1h),7.81(s,1h),6.73(d,j=4.3hz,1h),5.22(br s,1h),4.91(m,2h),4.41(dd,j=3.6,3.1hz,1h),3.62(m,2h),1.32(s,3h),1.23(s,3h);
13
c nmr(150mhz,dmso-d6):δ151.0,150.7,149.8,134.6,116.3,112.3,85.6,83.1,81.9,79.4,62.5,51.9,25.2,23.9.hrms(ei
+
)calcd for c
14h16
clin3o4[m+h]
+
451.9869;found 451.9888二醇d8a的相对立体化学的测定采用j-基构型分析确定了二醇d8a的相对立体化学。有关详细信息,请参见基于j的配置分析部分。
二醇d8b的相对立体化学的测定采用j-基构型分析确定了二醇d8b的相对立体化学。有关详细信息,请参见基于j的配置分析部分。核苷32的相对立体化学的测定核苷32的2d noesy分析支持了所示的立体化学。核苷33的相对立体化学的测定核苷33的2d noesy分析支持了所示的立体化学。二醇d8a对映体过量的测定按照常规步骤a和b,使用1:1的l-:d-脯氨酸的混合物,制备二醇d8a的外消旋品。使用ib柱通过手性hplc分离对映体二醇;洗脱剂:90:10(mecn:水)至10:90(mecn:水);检测波长230nm;保留时间:(+)-d8a为12.23min;(-)-d8a保留时间为13.39min。采用相同的方法(90:10e.r.)测定光学富集的ent-d8a二醇的对映体比例。二醇d8b对映体过量的测定按照常规步骤a和b,使用1:1的l-:d-脯氨酸的混合物,制备二醇d8b的外消旋品。使用ig柱通过手性hplc分离对映体二醇:洗脱剂:90:10(mecn:水)至10:90(mecn:水);检测波长230nm;保留时间:(-)-d8b为12.35min;(+)-d8b的保留时间为12.56min。使用相同的方法(93:7e.r.)测定光学富集的ent-d8b二醇的对映体比例。sm9、醛s9、羟醛缩合加合物a9、二醇加合物d9a/d9b和核苷类似物si9/na9的制备碘尿嘧啶(2.50g,10.5mmol,1.0当量),溴乙醛二乙基缩醛(1.91ml,12.7mmol,1.2当量)和碳酸钾(2.92g,21.1mmol,2.0当量)的溶液在dmf(70ml)中于90℃搅拌16小时。过滤
反应混合物,用200ml乙酸乙酯稀释滤液。有机层用水洗涤3次,分离,硫酸镁干燥,过滤,减压浓缩。通过闪蒸色谱法(戊烷:乙酸乙酯-75:25)纯化粗s9,得到白色固体s9(0.301g,产率8%)。s9溶液(0.142g,0.401mmol,1.0当量)在0.5m hcl(0.40ml)中加热至90℃并持续5小时。在完全转化为醛/水合物sm9后,将反应混合物减压浓缩,所得到的醛/水合物sm9用于反应,无需纯化。s9的数据:ir(neat):υ=2975,1686,1439,1121,1059,1021cm-1
;1h nmr(600mhz,cdcl3):δ8.56(br s,1h),7.82(s,1h),4.61(t,j=5.0hz),3.88(d,j=5.0hz),3.78(m,2h),3.54(m,2h),1.21(m,6h);
13
c nmr(150mhz,cdcl3):δ158.6,150.0,147.0(q,j=5.8hz),121.9(q,j=270.5hz),104.7(q,j=33.5hz),100.0,64.6,51.0,15.3.hrms(ei
+
)calcd for c
10h16
in2o4[m+h]
+
355.0149;found 355.0145顺式-和反式-氟醇a9的α-氟化/羟醛缩合和同步还原按照常规步骤a,在4℃,将s9(0.401mmol)、nfsi(0.126g,0.401mmol)、l-脯氨酸(0.046g,0.401mmol)和碳酸氢钠(0.034g,0.401mmol)的溶液在dmf(0.53ml)中搅拌12小时。然后加入二氧环己酮8(0.053ml,0.270mmol)的二氯甲烷(0.67ml),在4℃下搅拌反应混合物72小时。用闪蒸色谱法(戊烷-乙酸乙酯-1:1)纯化粗氟醇a9,得到黄色的油状氟醇a9。按照常规步骤b,在-15℃,将me4nhb(oac)3(0.066g,0.251mmol)和acoh(0.0.30ml,0.502mmol)加入到a9(0.021g,0.049mmol)在mecn(0.49ml)中的搅拌溶液中,并搅拌反应混合物24小时。由于稳定性和纯化方面的挑战,粗二醇d9a和d9b直接用于环化。二醇d9a和d9b的环化按照常规步骤c,d9a和d9b(16.2mg,0.038mmol,1当量)和2m氢氧化钠(0.038ml,0.38mmol,10当量)的溶液在mecn(1.51ml)中搅拌18小时。用闪蒸色谱法(ch2cl2:meoh-90:10)纯化粗核苷si9,得到白色固体核苷si9。将si9(10.3mg,0.025mmol)溶解于meod(0.25ml)中,并滴加和2滴1m盐酸,所得溶液在室温下放置12小时。随后,将反应混合物在减压下浓缩,得到白色固体na9。光谱数据与之前的报告相吻合(37)。核苷类似物si9的数据:1h nmr(600mhz,meod):δ7.99(s,1h),5.58(s,1h),4.35(d,j=4.5hz,1h),,4.19(dd,j=10.0,4.6hz,1h),4.08(dd,j=10.0,9.7hz,1h),3.83(m,2h),1.57(s,3h),1.45(s,3h);
13
c nmr(150mhz,meod):δ162.8,151.7,147.2,102.5,95.7,74.5,73.8,72.5,68.9,65.8,29.3,20.0
核苷na9的数据:[α]
d20
=-41(c=0.1,meoh);ir(neat):ν=3353,2929,1679,1447,1262,1101,1023,799cm
–1;1h nmr(600mhz,meod):δ8.61(s,1h),5.86(d,j=3.6hz,1h),4.16-4.17(m,2h),4.02-4.03(m,1h),3.89(dd,j=12.2,2.6hz,1h),3.76(dd,j=12.1,2.5hz,1h);
13
c nmr(150mhz,meod):δ162.8,152.2,147.3,90.9,86.3,76.1,70.9,68.3,61.7.hrms(ei
+
)calcd for c9h
12
in2o6[m+h]
+
370.9735;found:370.9739二醇d9a的相对立体化学的测定乙醇中的再结晶允许使用单x射线晶体学指定相对立体化学。核苷类似物36的制备向核苷类似物17(0.020g,0.083mmol,1.0当量)的干的二氯甲烷(0.83ml)溶液中加入tempo(1.3mg,0.008mmol,0.10当量)和(二乙酰氧基碘)苯((diacetoxyiodo)benzene)(0.067g,0.208mmol,2.5当量)。在18小时或通过1h nmr光谱监测17完全消耗之后,将反应混合物冷却至室温,并用二氯甲烷稀释。然后用饱和碳酸氢钠溶液洗涤有机层,用硫酸镁干燥,过滤,减压浓缩,得到粗产物36。通过闪蒸色谱法(戊烷:乙酸乙酯-1:1)纯化粗核苷36得到白色固体核苷36(0.019g,产率92%)。核苷类似物36的数据:[α]
d20
=-115.6(c 1.0in mecn);ir(neat):υ=3001,2989,1694,1374,1305,1088cm-1
;1h nmr(600mhz,cd3cn):δ7.80(d,j=2.4hz,1h),7.62(d,j=1.5hz,1h),6.36(dd,j=2.4,1.5hz,1h),5.78(s,1h),4.69(d,j=11.1hz,1h),4.22(d,j=10.0,5.0hz,1h),4.13(dd,j=10.6,10.6hz,1h),3.87(ddd,j=11.1,10.0,5.0hz,1h),1.56(s,3h),1.45(s,3h);
13
c nmr(150mhz,cd3cn):δ201.5,143.3,133.2,108.1,103.5,86.5,76.8,69.4,66.1,29.3,20.0.hrms(ei
+
)calcd for c
11h17
n2o5[m+h]
+
257.1132;found 257.1130核苷36的相对立体化学的测定
核苷36的2d noesy分析支持了所示的立体化学。核苷类似物37的制备向核苷类似物35(0.100g,0.352mmol,1当量)的thf(3.52ml)溶液中加入1,1
’‑
硫羰基二咪唑(1,1
’‑
thiocarbonyldiimidazole)(0.125g,0.704mmol,2当量)。将反应混合物搅拌24小时。随后,向反应混合物中加入二氯甲烷(10ml),然后水洗3次。所述有机层用硫酸镁干燥,过滤,减压浓缩,得到粗产物s37。用闪蒸色谱法(乙酸乙酯)纯化粗产物s37,得到s37(0.129g,96%)。核苷类似物s37的数据:[α]
d20
=+25.8(c 1.2in mecn);ir(neat):υ=3000,1701,1443,1375,1039,918,749cm-1
;1h nmr(600mhz,cd3cn):δ9.34(br s,1h),8.38(s,1h),7.73(s,1h),7.43(d,j=7.4hz,1h),7.04(s,1h),6.08(d,j=5.2hz,1h),5.88(d,j=5.2hz,1h),5.69(d,j=7.4hz,1h),4.22(m,2h),4.06(dd,j=10.4hz,1h),3.83(ddd,j=10.4,10.3,5.0hz,1h),1.55(s,3h),1.39(s,3h);
13
c nmr(150mhz,cd3cn):δ184.8,164.1,151.3,143.3,138.4,132.3,119.8,103.8,102.9,92.4,82.7,73.5,72.8,65.5,29.5,20.4.hrms(ei
+
)calcd for c
16h19
n4o6s[m+h]
+
395.1020;found 395.1010在氮气下,向核苷s37(0.020g,0.045mmol,1当量)在干燥的甲苯(3.0ml)溶液中加入三丁基锡氢化物(tributyltin hydride)(0.024ml,0.090mmol,2当量)和aibn(1.8mgs,0.011mmol,0.25当量)。将所得的反应混合物用氮气净化30分钟。随后,将反应混合物在90℃下搅拌16小时。用二氯甲烷(10ml)稀释反应混合物。有机层用水洗涤,分离,硫酸镁干燥,过滤,减压浓缩,得到粗产物37。用闪蒸色谱法(乙酸乙酯)纯化粗产物37,得到无色油状的核苷37(6.8mg,57%)。
核苷类似物37的数据:[α]
d20
=+7.8(c 0.32in meoh);1h nmr(600mhz,cd3cn):δ8.94(br s,1h),7.50(d,j=8.2hz,1h),6.14(dd,j=8.7,2.1hz,1h),5.63(d,j=8.2hz,1h),4.10(dd,j=10.0,4.6hz,1h),4.00(dd,j=10.3,10.0hz,1h),3.94(m,1h),3.35(ddd,j=10.3,10.0,4.6hz,1h),2.27(m,1h),2.17(m,1h),1.52(s,3h),1.37(s,3h);
13
c nmr(150mhz,cd3cn):δ164.1,151.6,142.6,103.3,102.2,84.4,76.3,72.7,65.6,36.4,29.8,20.5.hrms(ei
+
)calcd for c
12h17
n2o5[m+h]
+
269.1132;found 269.1111.核苷类似物38的制备在-78℃,向核苷36(0.020g,0.084mmol,1.0当量)的干的thf(0.84ml)溶液中加入甲基溴化镁(0.126ml,0.378mmol,4.5当量)。然后搅拌由此产生的反应混合物3.5小时。反应混合物在-78℃下用0.50ml的氯化铵:甲醇溶液(1:1-饱和氯化铵溶液:甲醇)淬灭,并加热至室温。用3ml的二氯甲烷稀释所得的混合物,然后用水洗涤两次。有机层用硫酸镁干燥,过滤,减压浓缩,得到粗产物38。用闪蒸色谱法(乙酸乙酯:戊烷-30:70)纯化粗产物38,得到白色固体核苷类似物38(19.1mg,90%)。核苷类似物38的数据:[α]
d20
=-117.7(c 0.57in ch2cl2);ir(neat):υ=3425,2992,1398,1384,1088,851cm-1
;1h nmr(600mhz,cd3cn):δ7.73(d,j=2.3hz,1h),7.60(d,j=1.3hz,1h),6.33(dd,j=2.3,1.3hz,1h),5.60(s,1h),4.13(d,j=10.0hz,1h),4.06(dd,j=9.8,4.7hz,1h),3.93(dd,j=10.1,9.8hz,1h),3.54(s,1h),3.48(ddd,j=10.1,10.0,4.7hz,1h),1.53(s,3h),1.41(s,3h),1.36(s,3h);
13
c nmr(150mhz,cd3cn):142.1,132.5,107.2,102.2,95.1,80.5,78.4,71.6,66.2,29.7,20.6,20.4.hrms(ei
+
)calcd for c
12h19
n2o4[m+h]
+
255.1339;found 255.1333核苷38的相对立体化学的测定
核苷38的2d noesy分析支持了所示的立体化学。核苷类似物39的制备在0℃下,向核苷类似物35(0.025g,0.088mmol,1当量)的二氯甲烷(0.45ml)溶液中,滴加三氟化二乙氨基硫(diethylaminosulfur trifluoride)(0.058ml,0.44mmol,5当量)。将反应混合物加热至室温并搅拌1小时。随后,加入乙酸乙酯(10ml),用饱和碳酸氢钠溶液洗涤有机层3次。然后将有机层进行分离、干燥、过滤和减压浓缩。用闪蒸色谱法(ch2cl2:meoh 95:5)纯化粗产物s39,得到白色固体2’,2
’‑
脱水尿(2’,2
’‑
anhydrouridine)s39(0.012g,产率51%)。然后将2’,2
’‑
脱水尿s39(0.011g,0.039mmol,1当量)溶解在1m hcl:meoh溶液(0.20ml:0.20ml)中。将反应混合物加热到50℃并持续24小时,然后减压浓缩,得到核苷39(9.5mg,产率100%)。光谱数据与之前的报告相吻合(41)。核苷类似物39的数据:1h nmr(600mhz,dmso-d6):δ11.28(d,j=2.1hz,1h),7.62(d,j=8.1hz,1h)5.98(d,j=4.5hz,1h),5.56(dd,j=8.1,2.1hz,1h),3.99(dd,j=4.4,3.2hz,1h),3.89(dd,j=3.6,3.2hz,1h),3.73(ddd,j=5.6,4.6,3.6hz,1h),3.60(dd,j=11.6,4.6hz,1h),3.56(dd,j=11.6,5.6hz,1h);
13
c nmr(150mhz,dmso-d6):δ163.4,150.5,142.3,100.0,85.1,84.7,75.5,75.1,60.7.hrms(ei
+
)calcd for c9h
13
n2o6[m+h]
+
245.0768;found 245.0777核苷类似物43的制备在-78℃,将甲基氯化镁(3.0m在thf中,1.49ml,4.47mmol,2.1当量)滴加到41(顺式-/反式-氟醇=3:1,1.00g,2.13mmol,1.0当量)的ch2cl2(10ml)溶液中。然后在该温度下将反应混合物搅拌2小时,然后逐渐加热至室温,并搅拌12小时。然后反应混合物用饱和氯化铵溶液淬灭,并用乙酸乙酯稀释。将有机层分离,硫酸镁干燥,过滤,减压浓缩。用闪蒸色谱法(ch2cl2中0-10%的甲醇)纯化粗产物43,得到白色固体核苷43(0.418克,42%)。核苷类似物43的数据:[α]
d20
=-13.6(c 0.28in ch2cl2);ir(neat):υ=3443,2250,1661,1053,1005,821cm-1
;1h nmr(600mhz,cdcl3):δ8.64(s,1h),7.55(s,1h),6.28(d,j=7.6hz,1h),4.92(ddd,j=9.8,7.5,4.4hz,1h),4.21(d,j=4.5hz,1h),3.83(d,j=12.6hz,1h),3.74(d,j=12.6hz,1h),3.40(d,j=9.8hz,1h),1.53(s,3h),1.49(s,3h),1.42(s,3h);
13
c nmr(150mhz,cdcl3):δ153.2,151.0,151.0,132.6,118.1,99.2,89.9,79.1,75.5,73.9,66.2,52.8,27.4,23.0,20.8.hrms(ei
+
)calcd for c
15h18
clin3o4[m+h]
+
466.0025;found 466.0054核苷43的相对立体化学的测定核苷43的2d noesy分析支持了所示的立体化学。核苷类似物44的制备在-78℃下,将甲基氯化镁(3.0m在thf中,1.56ml,4.68mmol,2.2当量)滴加到41(顺式-/反式-氟醇=3:1,1.00g,2.13mmol,1.0当量)的ch2cl2(20.0ml)溶液中。将得到的反应混合物在-78℃下搅拌5小时。然后反应混合物用氯化铵:甲醇溶液(1:1饱和氯化铵溶液:甲醇)淬灭并加热到室温。用二氯甲烷(50ml)稀释反应混合物,并将有机层分离,硫酸镁干燥,过滤,减压浓缩。用闪蒸色谱法(戊烷:乙酸乙酯-65:35)纯化粗产物42b,得到灰白色固体42b(0.498g,48%)。42b的数据:[α]
d20
=-17.7(c 1.8in ch2cl2);ir(neat):υ=3316,2991,1206,1086,863,736cm-1
;1h nmr(600mhz,dmso-d6):δ8.76(s,1h),8.28(s,1h),6.92(dd,j=45.8,3.3hz,1h),6.23(d,j=5.0hz,1h),4.65(s,1h),4.45(m,1h),3.44(d,j=11.1hz,1h),3.28(d,j=8.0hz,1h),3.23(d,j=11.1,1h),1.28(s,3h),1.13(s,3h),0.75(s,3h);
13
c nmr(150mhz,dmso-d6):δ151.5,151.4,151.2,134.3,116.0,98.3,90.2(d,j=202.7hz),74.1(d,j=4.5hz),70.1(d,j=25.1hz),70.0,66.7,55.2,28.4,19.7,18.1;
19
f nmr(470mhz,dmso-d6):δ
–
151.1.hrms(ei
+
)calcd for c
15h19
clfin3o4[m+h]
+
486.0087;found 486.0080向42b(0.100g,0.206mmol,1.0当量)在干mecn(2.0ml)的搅拌溶液中加入incl3(0.046g,0.206mmol,1.0当量)。将得到的反应混合物加热到50℃并持续2小时。然后添加2,2-二甲氧基丙烷(0.214mg,2.06mmol,10.0当量)和樟脑磺酸(9.6mg,0.041mmol,0.20当量),并在50℃下再将反应混合物搅拌1小时。然后浓缩反应混合物,并用闪蒸色谱法(0-10%甲醇在ch2cl2中)纯化,得到白色固体核苷44(0.049g,51%)。核苷类似物44的数据:[α]
d20
=+1.4(c 0.84in meod);1h nmr(600mhz,cdcl3):δ
8.58(s,1h),7.68(s,1h),6.83(d,j=4.5hz.1h),5.01(dd,j=6.0,4.7hz,1h),4.77(d,j=6.0hz,1h),3.79(dd,j=10.9,5.2hz,1h),3.74(dd,j=10.9,3.6hz,1h),2.02(dd,j=5.2,3.6hz,1h),1.48(s,3h),1.41(s,3h),1.31(s,3h);
13
c nmr(150mhz,cdcl3):δ152.6,150.8,150.3,134.5,117.4,113.2,85.1,85.0,83.0,81.1,69.5,50.8,25.6,24.1,17.4.hrms(ei
+
)calcd for c
15h18
clin3o4[m+h]
+
466.0025;found 466.0000核苷44的相对立体化学的测定核苷44的2d noesy分析支持了所示的立体化学。核苷类似物45的制备在-78℃,将乙基氯化镁(0.5m在thf中,8.94ml,4.47mmol,2.1当量)滴加到41(顺式-/反式-氟醇=3:1,1.00g,2.13mmol,1.0当量)的二氯甲烷(10ml)溶液中。将反应混合物在此温度下搅拌2小时,然后逐渐加热至室温,并搅拌12小时。然后将反应混合物用饱和氯化铵溶液淬灭,并用乙酸乙酯稀释。将有机层分离,硫酸镁干燥,过滤,减压浓缩。用闪蒸色谱法(ch2cl2中0-10%甲醇)纯化粗产物45得到白色固体核苷45(0.415克,41%)。核苷类似物45的数据:[α]
d20
=-29.5(c 0.58in meoh);ir(neat):υ=3291,2924,1446,1201,1023,600cm-1
;1h nmr(600mhz,dmso-d6):δ8.72(s,1h),8.02(s,1h),6.44(d,j=8.1hz,1h),5.05(dd,j=8.1,3.6hz,1h),4.44(d,j=3.6hz,1h),4.16(s,1h),4.01(d,j=13.2hz,1h),3.82(d,j=13.2hz,1h),3.44(br s,1h),1.49(s,3h),1.43(s,3h);
13
c nmr(150mhz,dmso-d6):δ151.7,151.4,151.1,132.8,116.6,97.5,86.5,81.1,80.5,75.0,74.1,72.3,64.2,53.0,28.5,18.9.hrms(ei
+
)calcd for c
16h16
clin3o4[m+h]
+
475.9869;found 475.9849核苷45的相对立体化学的测定相对立体化学是基于异聚质子与化合物43和46的化学位移的比较而确定的。核苷类似物46的制备在-78℃,将苯基氯化镁(2.0m在thf中,2.24ml,4.47mmol,2.1当量)滴加到41(顺式-/反式氟醇=3:1,1.00g,2.13mmol,1.0当量)的ch2cl2(10ml)溶液中。将反应混合物在此温度下搅拌2小时,然后逐渐加热至室温,并搅拌12小时。然后将反应混合物用饱和氯化铵溶液淬灭,并用乙酸乙酯稀释。将有机层分离,硫酸镁干燥,过滤,减压浓缩。用闪蒸色谱法(ch2cl2中0-10%甲醇)纯化粗产物46,得到白色固体核苷46(0.496克,45%)。
核苷类似物46的数据:[α]
d20
=-23.6(c 1.7in ch2cl2);ir(neat):υ=3309,2990,2938,1575,1538,1445,1200cm-1
;1h nmr(600mhz,dmso-d6):δ8.70(s,1h),7.63(s,1h),7.43(m,5h),6.55(d,j=8.3hz,1h),5.55(d,j=6.9hz,1h),4.77(d,j=3.8hz,,1h),4.67(ddd,j=8.3,6.9,3.8hz,1h),3.81(d,j=12.9hz,1h),3.68(d,j=12.9hz,1h),1.62(s,3h),1.50(s,3h);
13
c nmr(150mhz,dmso-d6):δ152.0,151.3,151.0,140.4,133.4,128.5,128.0,125.3,111.8,97.4,86.1,80.8,73.9,72.5,67.0,54.3,28.3,20.2.hrms(ei
+
)calcd for c
20h20
clin3o4[m+h]
+
528.0182;found 528.0206.核苷46的相对立体化学的测定核苷46的2d noesy分析支持了所示的立体化学。核苷类似物47的制备在-78℃,将乙炔基氯化镁(0.5m在thf中,8.94ml,4.47mmol,2.1当量)滴加到41(顺式-/反式-氟醇=3:1,1.00g,2.13mmol,1.0当量)的ch2cl2(20ml)溶液中。将得到的反应混合物在-78℃下搅拌1小时。然后将反应混合物用氯化铵:甲醇溶液(1:1-饱和氯化铵溶液:甲醇)猝灭并加热到室温。用ch2cl2(50ml)稀释反应混合物,分离有机层,硫酸镁干燥,过滤,减压浓缩。用闪蒸色谱法(戊烷:乙酸乙酯-65:35)对粗产物s47进行纯化,得到灰白色固体s47(0.720g,68%,1:1的非对映体混合物)。向s47(0.050g,0.101mmol,1.0当量)在干式mecn(2.0ml)的搅拌溶液中加入incl3(0.022g,0.101mmol,1.0当量)。将得到的反应混合物加热到50℃并持续2小时。当2,2-二甲氧基丙烷(0.124ml,1.01mmol,10.0当量)和樟脑磺酸(4.7mg,0.020mmol,0.20当量)加入后,将反应混合物在50℃下进一步搅拌1小时。然后将反应混合物浓缩并用闪蒸色谱法(0-10%甲醇在ch2cl2中)进行纯化,得到白色固体核苷47(0.029g,60%)。
[0001]
核苷类似物47的数据:[α]
d20
=+6.3(c 2.0in ch2cl2);1h nmr(600mhz,cdcl3):δ
8.59(s,1h),7.82(s,1h),6.85(d,j=4.6hz,1h),5.03(dd,j=6.0,4.9hz,1h),4.98(d,j=6.0hz,1h),3.97(dd,j=11.5,4.4hz,1h),3.92(dd,j=11.5,3.5hz,1h),2.82(s,1h),2.18(dd,j=4.4,3.5hz,1h),1.53(s,3h),1.34(s,3h);
13
c nmr(150mhz,cdcl3):δ152.7,150.9,1505.,134.6,117.4,114.6,85.3,83.0,82.9,80.6,78.2,77.8,68.7,51.4,25.7,24.5.hrms(ei
+
)calcd for c
16h16
clin3o4[m+h]
+
475.9869;found 475.9885核苷47的相对立体化学的测定核苷47的2d noesy分析支持了所示的立体化学。核苷类似物48的制备在-78℃,将甲基碘化镁(3.0m在thf中,0.39ml,1.16mmol,3当量)滴加到a5(0.100g,0.388mmol,1当量)的ch2cl2溶液中。将所得到的反应混合物逐渐加热至-10℃,然后搅拌2小时。通过薄层色谱分析监测反应完成后,将反应混合物用饱和氯化铵溶液淬灭,并用ch2cl2稀释。有机层随后可以用水洗两次,也可以用盐水洗一次。然后有机层依次用mgso4干燥、过滤、减压浓缩。然后用闪蒸色谱法(戊烷:乙酸乙酯-25:75)纯化粗产品s48,得到黄色透明油状液体s48(0.089g,84%)。s48的数据:1h nmr(600mhz,cdcl3):δ8.16,8.02,7.76,7.76,6.80,6.58,4.62,4.52,4.40,4.31,4.07,3.81,3.59,3.55,3.45,3.25,3.13,3.10,1.52,1.47,1.45,1.40,1.38,1.17;
13
c nmr(150mhz,cdcl3):δ134.2,134.1,124.9,124.3,99.8,99.8,95.6,93.4,72.4,72.4,71.9,71.8,70.2,70.0,67.9,67.8,28.8,28.7,20.0,19.8,19.2,18.5;
19
f nmr(470mhz,cdcl3):δ
–
157.8,-162.8hrms(ei
+
)calcd for c
11h19
fn3o4[m+h]
+
276.1354;found 276.1366向s48的(0.060g,0.218mmol,1当量)干燥的mecn(2.18ml)溶液中加入sc(otf)3(0.268g,0.545mmol,2.5当量)。搅拌反应混合物16小时后,向反应混合物中加入0.50ml乙酸酐和0.50ml的吡啶。将反应混合物再搅拌4小时,然后用ch2cl2稀释。有机层用1m hcl洗涤2次、水洗涤一次,用硫酸钠干燥,过滤,减压浓缩,得到粗产物48。通过闪蒸色谱法(戊烷:乙酸乙酯-60:40)纯化粗产物48,得到48(0.024g,产率32%)。
核苷类似物48的数据:[α]
d20
=+18.4(c 1.46in ch2cl2);ir(neat):υ=2925,1744,1374,1215,1049cm-1
;1h nmr(600mhz,cdcl3):δ7.76(d,j=0.60hz,1h),7.75(d,j=0.60hz,1h)6.19(d,j=4.7hz,1h),6.02(dd,j=5.4,4.7hz,1h),5.67(d,j=5.4hz,1h),4.17(d,j=12.0hz,1h),4.08(d,j=12.0hz,1h),2.15(s,3h),2.09(s,3h),2.03(s,3h),1.37(s,3h);
13
c nmr(150mhz,cdcl3):δ170.3,169.3,169.2,134.4,122.7,89.4,85.6,75.0,71.9,67.9,20.8,20.5,20.5,19.3.hrms(ei
+
)calcd for c
14h20
n3o7[m+h]
+
342.1296;found 342.1312核苷类似物48的相对立体化学的测定核苷48的2d noesy分析支持了所示的立体化学。核苷类似物49的制备按照常规步骤e,在-78℃,将对甲苯基溴化镁(p-tolylmagnesium bromide)(1.0m在thf中,0.712ml,0.71mmol)加入到59(0.050g,0.158mmol)的二氯甲烷(6.30ml)溶液中。反应混合物搅拌4.5小时。无需进一步纯化,将粗产物s49溶解在mecn(1.58ml)中,并加入2m氢氧化钠(0.198ml,0.395mmol),然后将反应混合物加热至50℃并持续4小时。用闪蒸色谱法(戊烷:乙酸乙酯-35:65)对粗产物49进行纯化,得到无色油状的核苷49(0.024g,两步产率39%)。核苷类似物49的数据:[α]
d20
=-56.5(c 0.4in meoh);ir(neat):υ=3432,2939,1700,1466,1378,1129,1051cm-1
;1h nmr(600mhz,cd3cn):δ8.96(br s,1h),7.38(d,j=
8.1hz,2h),7.26(d,j=8.1hz,2h),6.78(d,j=0.90hz,1h),6.24(d,j=8.2hz,1h),4.76(d,j=3.8hz,1h),4.19(s,1h),3.80(d,j=13.2hz,1h),3.73(d,j=13.2hz,1h),3.48(br s),2.35(s,3h),1.68(d,j=0.90hz,3h),1.60(s,3h),1.49(s,3h);
13
c nmr(150mhz,cd3cn):δ164.6,152.8,139.6,138.7,137.4,130.6,126.7,112.0,99.2,88.9,81.6,74.9,74.3,68.7,29.0,21.4,20.8,12.8.hrms(ei
+
)calcd for c
20h25
n2o6[m+h]
+
389.1707;found 389.1707核苷49的相对立体化学的测定核苷49的2d noesy分析支持了立体化学结果核苷类似物50的制备按照常规步骤e,在-78℃,将环丙基溴化镁(cyclopropylmagnesium bromide)(1.0m在2-甲基四氢呋喃中,0.79ml,0.79mmol,5当量)加入到59(0.050g,0.158mmol,1当量)的ch2cl2(6.30ml)溶液中。然后搅拌反应混合物5小时。无需进一步纯化,直接将粗产物s50溶解在mecn(1.60ml)中,加入2m氢氧化钠(0.193ml,0.395mmol),然后在50℃下搅拌反应混合物4小时。用闪蒸色谱法(戊烷:乙酸乙酯-30:70)对粗产物50进行纯化,得到灰白色固体核苷50(0.021g,产率40%)。核苷类似物50的数据:[α]
d20
=-32.6(c 0.47in ch2cl2);ir(neat):υ=3500,32512997,2175,1690,1088,888cm-1
;1h nmr(600mhz,cdcl3):δ7.10(s,1h),6.04(d,j=7.9hz,1h),4.25(dd,j=7.9,5.1hz.1h),4.08(d,j=5.1hz,1h),3.70(d,j=11.9hz,1h),3.63(d,j=11.9hz,1h),3.15(br s,1h),1.93(s,3h),1.44(s,3h),1.43(s,3h),1.21(m,1h),0.63(m,1h),0.55(m,1h),0.46(m,1h),0.42(m,1h);
13
c nmr(150mhz,cdcl3):δ163.3,151.0,134.9,111.9,100.1,87.5,81.2,74.0,72.5,64.3,25.9,25.6,16.2,12.9,1.31,0.50.hrms(ei
+
)calcd for c
16h22
n2o6[m+h]
+
339.1551;found 339.1575核苷50的相对立体化学的测定
核苷50的2d noesy分析支持了所示的立体化学。核苷类似物51的制备按照常规般步骤e,在-78
°
,将对甲氧基苯基溴化镁(0.5m在thf中,1.58ml,0.79mmol,5当量)加入到59(0.050g,0.158mmol,1当量)的ch2cl2(6.30ml)溶液中,并搅拌反应混合物5小时。无需进一步纯化,直接将粗产物s51溶解在mecn(1.60ml)中,并加入2m氢氧化钠(0.193ml,0.395mmol),在50℃下搅拌反应混合物4小时。用闪蒸色谱法(戊烷:乙酸乙酯-30:70)对粗产物51进行纯化,得到白色固体核苷51(0.026g,产率41%)。核苷类似物51的数据:[α]
d20
=-52.8(c 1.0in ch2cl2);ir(neat):υ=3197,2990,1693,1252,1036,834cm-1
;1h nmr(600mhz,cdcl3):δ7.38(d,j=8.7hz,2h),6.96(d,j=8.7hz,2h),6.78(s,1h),6.37(d,j=7.9hz,1h),4.75(d,j=4.1hz,1h),4.16(m,1h),3.87(d,j=13.1hz,1h),3.84(s,3h),3.79(d,j=13.1,1h),2.99(br s,1h),1.63(s,3h),1.56(s,3h);
13
c nmr(150mhz,cdcl3):δ163.2,159.9,151.1,135.8,131.8,126.5,114.5,111.7,98.7,88.7,80.6,74.8,73.2,67.7,55.6,28.1,20.4,12.7.hrms(ei
+
)calcd for c
20h25
n2o7[m+h]
+
405.1656;found 405.1650核苷类似物51相对立体化学的测定
核苷51的2d noesy分析支持了所示的立体化学。核苷类似物52的制备按照常规步骤e,在-78℃,将对甲氧基苯基溴化镁(0.5m在thf中,4.66ml,2.33mmol,3当量)加入到a1(0.200g,0.775mmol,1当量)的二氯甲烷(7.75ml)溶液中,搅拌反应混合物6小时。用闪蒸色谱法(乙酸乙酯-戊烷-4:6)纯化粗产物s52,得到s52(0.157g,收率55%)。将s52(0.155g,0.423mmol,1当量)溶解在mecn(2.82ml)中,并加入2m氢氧化钠(0.53毫升,1.06mmol,2.5当量),所得反应混合物在50℃下搅拌5小时。用闪蒸色谱法(戊烷:乙酸乙酯-40:60)纯化粗核苷类似物52,得到浅橙色油状的52(0.085g,产率58%)。核苷类似物52的数据:[α]
d20
=-14.8(c 1.4in ch2cl2);ir(neat):υ=3418,2991,1611,1512,1250,1032,759cm-1
;1h nmr(600mhz,cd3cn):δ7.69(d,j=2.7hz,1h),7.56(d,j=1.4hz,1h),7.39(d,j=8.9hz,2h),6.91(d,j=8.9hz,2h),6.35(dd,j=2.7,1.4hz,1h),5.99(d,j=7.9hz,1h),4.73(dd,j=7.9,3.7hz,1h),4.59(d,j=3.7hz,1h),3.92(d,j=13.3hz,1h),3.78(s,3h),3.68(d,j=13.3hz,1h),1.62(s,3h),1.51(s,3h);
13
c nmr(150mhz,cd3cn):δ160.6,141.5,133.7,132.1,128.4,114.8,107.6,99.1,93.9,82.0,75.9,75.1,68.9,56.3,29.0,21.2.hrms(ei
+
)calcd for c
18h23
n2o5[m+h]
+
347.1601;found 347.1610核苷52的相对立体化学的测定
核苷52的2d noesy分析支持了所示的立体化学。核苷类似物53的制备按照常规步骤e,在-78℃,将甲基溴化镁(3.0m在thf中,0.258ml,0.78mmol,4当量)加入a1(0.050g,0.194mmol,1当量)的二氯甲烷(3.90ml)溶液中。搅拌反应混合物6小时。用闪蒸色谱法(乙酸乙酯-戊烷-6:4)纯化粗产物s53,得到s53(0.026g,收率49%)。将s53(0.030g,0.109mmol)溶解于mecn(1.09ml)中,并加入2m氢氧化钠(0.545ml,1.09mmol,10当量),所得反应混合物在50℃下搅拌5小时。用闪蒸色谱法(戊烷:乙酸乙酯-25:75)纯化粗核苷类似物53,纯化得到浅黄色油状的53(0.017g,产率61%)。核苷类似物53的数据:[α]
d20
=+11.3(c 0.38in ch2cl2););ir(neat):υ=3383,2992,2922,1382,1199,1090,908cm-11
h nmr(600mhz,cdcl3):δ7.60(d,j=2.4hz,1h),7.59(d,j=1.6hz,1h),6.35(dd,j=2.4,1.6hz,1h),5.29(d,j=1.3hz,1h),4.12(dd,j=3.0,1.3hz,1h),3.98(d,j=3.0hz,1h),3.76(d,j=11.3hz,1h),3.52(d,j=11.3hz,1h),1.47(s,3h),1.44(s,3h),1.41(s,3h);
13
c nmr(150mhz,cdcl3):δ141.3,129.3,107.4,99.6,72.6,70.3,67.3,64.9,57.0,28.8,20.5,19.0.hrms(ei
+
)calcd for c
12h19
n2o4[m+h]
+
255.1339;found 255.1320核苷类似物53的相对立体化学的测定核苷53的2d noesy分析支持了所示的立体化学。核苷类似物54的制备在0℃,将对氯苯溴化镁(p-chlorophenylmagnesium bromide)(1.0m在乙醚中,4.32ml,4.32mmol,3.2当量)滴加入氟醇羟醇缩合加合物a6(0.500g,1.35mmol,1当量)在thf(10.0ml)中的搅拌溶液中。然后将得到的反应混合物在室温下搅拌14小时,并在40℃下
再搅拌8小时。然后用乙酸乙酯(100ml)稀释反应混合物,并用水(100ml)和盐水(50ml)各洗涤一次。分离有机层,并用硫酸镁干燥,过滤,减压浓缩,得到粗产物54。用闪蒸色谱法(戊烷:乙酸乙酯
–
50:50)纯化粗核苷类似物54得到54(0.289克,46%)。核苷54的数据:[α]
d20
=+10.5(c 0.8in ch2cl2);ir(neat):υ=3087,2996,1699,1467,1283,1129,1085cm-1
;1h nmr(600mhz,dmso-d6):δ11.94(br s,1h),8.74(s,1h),7.57(d,j=8.7hz,2h),7.50(d,j=8.7hz,2h),6.13(d,j=7.2hz,1h),5.67(br s,1h),4.66(d,j=4.3hz,1h),4.17(dd,j=6.8,4.3hz,1h),3.98(d,j=13.4hz,1h),3.88(d,j=13.4hz,1h),1.63(s,3h),1.409s,3h);
13
c nmr(150mhz,cd3cn):δ159.9,150.5,144.3(q,j=5.9hz),138.0,134.9,129.9,128.1,124.1(q,j=269.0hz),104.0(q,j=32.0),99.6,84.7,81.6,73.6,73.6,67.7,28.6,19.9;
19
f nmr(470mhz,cd3cn):δ
–
62.9.hrms(ei
+
)calcd for c
19h19
clf3n2o6[m+h]
+
463.0878;found 463.0875核苷54的相对立体化学的测定核苷54的2d noesy分析支持了所示的立体化学。核苷类似物57的制备向核苷35(0.285g,1.0mmol,1.0当量)在干燥的二氧烷(20ml)的溶液中加入(二乙氧基碘)苯((diacetoxyiodo)benzene)(0.805g,2.5mmol,2.5当量)和tempo(0.031g,0.20mmol,0.2当量)。然后在室温下搅拌反应混合物24小时,直到薄层色谱分析检测原料完全消耗。将反应混合物浓缩至2ml,用闪蒸色谱法(ch2cl2:et2o-75:25)纯化,得到白色固体酮56(0.265g,0.94mmol,产率94%)。将酮56(0.053g,0.19mmol,1.0当量)溶解在甲醇(0.94ml)中,并加入3滴accl。在室温下搅拌溶液12小时,直到薄层色谱分析检测原料完全消耗。将反应混合物在减压条件下浓缩成白色固体s57。光谱数据与之前的报告(50)相匹配。粗产物随后溶解在四氢呋喃(4.0ml)中,并将所得溶液冷却到-78℃,然后加入甲基溴化镁(3.0m在thf中,0.38ml,1.13mmol,6.0当量)。将得到的棕色悬浮液在-78℃下搅拌3小时。然后将反应混合物在-78℃下用甲醇:tfa(10:1)溶液淬灭,然后进行减压浓缩。用闪蒸色谱法(ch2cl2:meoh-85:15)纯化粗产物57,得到白色固体核苷类似物(0.024g,产率49%)。光谱数据与之前的报告(51)相符。
核苷类似物57的数据:1h nmr(600mhz,meod):δ7.86(d,j=8.1hz,1h),5.96(s,1h),5.64(d,j=8.1hz,1h),3.85(m,4h),1.29(s,3h).hrms(ei
+
)calcd for c
10h15
n2o6[m+h]
+
259.0925;found 259.0915核苷类似物60的制备向59(0.100g,0.316mmol,1当量)在thf(3.10ml)的搅拌溶液中加入bnnh2(0.086ml,0.790mmol,2.5当量)和冰醋酸(18.2μl,0.316mmol,1当量),然后将所得混合物在20℃下搅拌1小时。然后加入nabh3cn(0.050g,0.79mmol,2.5当量),并将混合物再搅拌一小时。然后用二氯甲烷将反应混合物稀释至0.05m,然后用水处理。分离各层,有机层用盐水洗涤,硫酸镁干燥,减压浓缩。使用所得产物s60而不进行任何进一步纯化。向s60在mecn(8.7ml)的搅拌溶液中加入2m氢氧化钠(0.240ml,0.478mmol,1.1当量)。反应混合物在室温下搅拌14小时。然后用二氯甲烷稀释反应混合物,用饱和氯化铵溶液淬灭。有机层用饱和氯化铵溶液和水洗涤,用硫酸镁干燥,过滤,减压浓缩。用闪蒸色谱法(乙酸乙酯:戊烷-80:20)纯化粗产物60,得到浅黄色油状的核苷类似物60(0.060g,两步产率49%)。核苷类似物60的数据:[α]
d20
=-15.5(c 0.53in ch2cl2);ir(neat):υ=2990,1670,1382,1200,1078,701cm-1
;1h nmr(600mhz,cdcl3):δ7.23-7.32(m,4h),7.19(d,j=7.0hz,2h),5.07(s,1h),4.11(d,j=4.8hz,1h),3.81(d,j=12.9hz,1h),3.77(d,j=12.9hz,1h),3.72(dd,j=10.4,4.6hz,1h),3.67(dd,j=10.4,10.2hz,1h),3.61(dd,j=9.8,4.8hz,1h),3.11(ddd,j=10.2,9.8,4.6hz,1h),1.86(s,3h),1.49(s,3h),1.46(s,3h);
13
c nmr(150mhz,cdcl3):δ163.5,150.7,136.8,135.9,129.1,128.7,128.2,110.1,101.0,83.1,74.9,73.2,66.6,58.1,58.0,29.2,19.9,12.8.hrms(ei
+
)calcd for c
20h26
n3o5[m+h]
+
388.1867;found 388.1843.核苷60的相对立体化学的测定
核苷60的2d noesy分析支持了所示的立体化学。核苷类似物61的制备按照常规步骤e,在-78℃,将烯丙基溴化镁(1.0m在乙醚中,1.42ml,1.42mmol,4.5当量)加入到59(0.100g,0.316mmol,1当量)的二氯甲烷(12.6ml)溶液中。然后搅拌反应混合物5小时。在没有进一步纯化的情况下,将粗产物s61溶解在mecn(3.16ml)中,并加入2m氢氧化钠(0.395ml,0.79mmol,2.5当量)。然后反应混合物在50℃下搅拌4小时。用闪蒸色谱法(ch2cl2:meoh-4:96)对粗产物61进行纯化,得到暗橙色油状的核苷类似物61(0.050g,产率47%)。核苷类似物61的数据:[α]
d20
=-6.0(c 0.4in meoh);ir(neat):υ=3340,2992,1670,1376,1044cm-1
;1h nmr(600mhz,cd3cn):δ8.95(br s,1h),7.27(s,1h),6.02(d,j=8.3hz,1h),5.87(m,1h),5.22(d,j=17.7hz,1h),5.20(d,j=10.1hz,1h),4.31(ddd,j=9.3,8.3,4.9hz,1h),4.11(d,j=4.9hz,1h),3.68(d,j=12.2hz,1h),3.64(d,j=12.2hz,1h),3.41(d,j=9.3hz,1h),2.50(dd,j=14.2,6.7hz,1h),2.41(dd,j=14.2,8.1hz,1h),1.85(s,3h),1.40(s,3h),1.39(s,3h);
13
c nmr(150mhz,cd3cn):δ164.6,152.3,136.8,133.7,120.4,112.3,100.3,88.4,81.7,73.9,73.3,65.3,41.7,26.9,22.4,12.8.hrms(ei
+
)calcd for c
16h22
n2o6[m+h]
+
339.1551;found 339.1556核苷61的相对立体化学的测定核苷61的2d noesy分析支持了所示的立体化学。核苷类似物62的制备向核苷61(0.022g,0.061mmol,1当量)在干thf(0.61ml)的溶液中加入1,1'-硫羰基二咪唑(0.022g,0.122mmol,2当量)。然后搅拌反应混合物18h。随后加入ch2cl2(5ml),并
用水洗涤3次。有机层用硫酸镁干燥,过滤,减压浓缩,得到粗产物s62。用闪蒸色谱法(戊烷:乙酸乙酯-40:60)纯化粗产物s62得到s62(0.018g,产量为66%)。在氮气下,向核苷s62(0.014g,0.031mmol,1当量)在干燥的甲苯(4.45ml)的溶液中加入三丁基锡氢化物(8.35μl,0.031mmol,1当量)和aibn(5.1mgs,0.031mmol,1.0当量)。然后将所得的反应混合物用氮气净化30分钟。随后,将反应混合物在90℃下搅拌16小时。在竞争中(upon competition),在向反应混合物中加入二氯甲烷并用水洗涤。有机层用硫酸镁干燥,过滤,减压浓缩,得到粗产物62。用闪蒸色谱法(乙酸乙酯)对粗核苷类似物62进行纯化,得到白色固体核苷类似物62(6.0mg,61%)。核苷类似物62的数据:[α]
d20
=+13.3(c 0.46in ch2cl2);ir(neat):υ=2924,1690,1467,1375,1263,1226,1053cm-1
;1h nmr(600mhz,cdcl3):δ8.26(s,1h),7.31(d,j=1.1hz,1h),6.38(dd,j=9.6,4.8hz,1h),5.86(m,1h),5.25-5.27(m,2h),4.22(d,j=5.2hz,1h),3.69(d,j=12.0hz,1h),3.64(d,j=12.0hz,1h),2.50(m,2h),2.41(dd,j=13.5,4.8hz,1h),2.00(dd,j=13.5,9.6,5.2hz,1h),1.92(s,3h),1.37(s,6h);
13
c nmr(150mhz,cdcl3):δ163.3,150.0,135.0,131.9,120.2,111.1,99.5,85.8,84.0,73.9,63.9,40.9,37.8,25.6,22.5,12.7hrms(ei
+
)calcd for c
16h23
n2o5[m+h]
+
323.1601;found 323.1580氟醇63和64的制备按照常规步骤e,在-78℃,将乙炔基氯化镁(0.5m在thf中,3.5ml,1.75mmol,3.5当量)加入到59(0.160g,0.50mmol,1当量)在二氯甲烷(25.0ml)的溶液中。然后搅拌反应混合物4小时。通过闪蒸色谱法(乙酸乙酯:己烷-70:30)纯化粗产物63和64,得到白色固体63(0.072g,产率42%)和64(0.058g,34%)。氟醇63的数据:[α]
d20
=-60.8(c 0.4in meoh);ir(neat):υ=3320,2944,2832,1670,1449,1022,638cm-1
;1h nmr(600mhz,dmso-d6):δ11.47(br s,1h),7.56(s,1h),6.36(dd,j=43.7,4.1hz,1h),6.21(d,j=5.3hz,1h),5.37(br s,1h),4.14(m,1h),3.71(d,j=8.7hz,1h),3.68(br s,1h),3.42(s,1h),3.16(d,j=5.0hz,1h),1.78(s,3h),1.33(s,3h),1.21(s,3h);
13
c nmr(150mhz,dmso-d6):δ163.6,150.0,136.7,109.2,98.8,92.7(d,j=206.6hz),83.7,76.2,72.8(d,j=2.8hz),71.2(d,j=24.6hz),68.2,65.7,27.8,18.7,12.1;
19
fnmr(470mhz,dmso-d6):δ
–
170.5hrms(ei
+
)calcd for c
15h20
n2o6[m+h]
+
343.1300;found 343.1298.
氟醇64的数据:[α]
d20
=-38.0(c 1.2inmeoh);ir(neat):υ=ir(neat):υ=3395,2994,1694,1468,1381,1282,1043cm-1
;1h nmr(600mhz,cd3cn):δ9.29(br s,1h),7.41(s,1h),6.40(dd,j=43.4,4.6hz,1h),4.54(m,1h),4.27(m,1h),4.22(m,1h),3.82(d,j=9.5hz,1h),3.79(d,j=11.5hz,1h),3.75(d,j=11.5hz,1h),2.81(s,1h),1.85(s,3h),1.41(s,3h),1.28(s,3h);
13
c nmr(150mhz,cd3cn):δ164.8,151.4,137.7,111.5,100.8,94.1(d,j=206.9hz),84.4,75.7,73.6(d,j=3.8hz),73.4(d,j=24.7hz),69.3,67.3,28.8,19.4,12.8;
19
f nmr(470mhz,cd3cn):δ
–
175.5hrms(ei
+
)calcd for c
15h20
n2o6[m+h]
+
343.1300;found 343.1305核苷类似物65的制备按照常规步骤c,将63(0.100g,0.292mmol,1.0当量)和氢氧化钠(29.2mg,0.73mmol,2.5等量)的溶液在mecn(2.0ml)中加热到50℃并持续36小时。用闪蒸色谱法(0-10%的meoh在二氯甲烷中)对粗产物65进行纯化,得到白色粉末核苷类似物65(58.6mg,产率62%)。核苷类似物65的数据:[α]
d20
=-8.7(c 0.6in ch2cl2);ir(neat):υ=2994,1748,1690,1270,1043cm-1
;1h nmr(600mhz,dmso-d6):δ11.42(s,1h),7.61(d,j=1.3hz,1h),5.46(s,1h),4.86(s,1h),4.63(d,j=11.2hz,1h),4.45(d,j=2.6hz,1h),4.37(d,j=2.6hz,1h),4.23(d,j=11.2hz,1h),3.91(s,1h),1.84(s,3h),1.51(s,3h),1.30(s,3h);
13
c nmr(150mhz,dmso-d6):δ163.8,158.8,150.0,135.0,109.3,100.4,87.2,83.0,78.4,76.5,71.9,58.5,28.7,19.5,12.0hrms(ei
+
)calcd for c
15h19
n2o6[m+h]
+
323.1238;found 323.1235核苷类似物68的制备按照常规c,将64(0.220g,0.64mmol,1当量)和2m氢氧化钠(0.640ml,1.28mmol,2.0当量)的溶液在mecn(6.4ml)中加热至50℃并搅拌24小时。用闪蒸色谱(meoh:ch2cl
2-3:97)对粗产物66进行纯化,得到白色粉末核苷类似物66(0.144mg,产率70%)。
核苷类似物66的数据:[α]
d20
=+30.8(c 1.66in ch2cl2);1h nmr(600mhz,cd3cn):δ9.06(br s,1h),7.48(s,1h),6.16(d,j=8.2hz,1h),4.61(ddd,j=8.4,8.2,3.7hz,1h),4.41(d,j=3.7hz,1h),4.06(d,j=13.3hz,1h),3.88(d,j=13.3hz,1h),3.64(d,j=8.4hz,1h),3.29(s,1h)1.86(s,3h),1.48(s,3h),1.43(s,3h);
13
c nmr(150mhz,cd3cn):δ164.7,152.5,136.9,112.7,99.3,89.4,81.2,80.8,76.5,75.5,73.9,65.9,29.1,19.7,13.1.hrms(ei
+
)calcd for c
15h19
n2o6[m+h]
+
323.1238;found 323.1245核苷66的相对立体化学的测定核苷66的2d noesy分析支持了所示的立体化学。将66(0.050g,0.155mmol,1当量)在干燥的二氯甲烷(0.78ml)的溶液冷却到0℃,然后在5分钟内逐滴添加三氟化二乙氨基硫(0.102ml,0.776mmol,5当量)。将所得反应混合物在3小时缓慢加热到室温。通过薄层色谱分析监测反应完成后,用5ml乙酸乙酯稀释反应混合物,用3ml水(3倍)洗涤。随后,有机层经过硫酸镁干燥处理,过滤,并在减压浓缩。用闪蒸色谱法(乙酸乙酯)纯化粗产物,得到白色固体核苷类似物s66(0.043g,91%)。核苷类似物s66的数据:[α]
d20
=-47.5(c 1.1in mecn);ir(neat):υ=3284,3002,1626,1554,1497,1134,1066,1030cm-1
;1h nmr(600mhz,cd3cn):δ7.46(s,1h),6.32(d,j=5.3hz,1h),5.13(d,j=5.3hz,1h),4.74(s,1h),4.10(d,j=13.7hz,1h),4.00(d,j=13.7hz,1h),2.87(s,1h),1.87(s,3h),1.47(s,3h),1.34(s,3h);
13
c nmr(150mhz,cd3cn):δ173.1,161.5,132.3,119.6,99.7,91.8,87.4,79.8,79.1,77.8,74.6,64.9,29.0,19.3,14.4.hrms(ei
+
)calcd for c
15h17
n2o5[m+h]
+
305.1132;found 305.1108向s66(0.042g,0.138mmol,1当量)在湿mecn(2.76ml)的溶液中加入incl3(0.122g,0.553mmol,4当量)。将得到的反应混合物加热到50℃,并搅拌16小时或用薄层色谱法检测反应完成。然后将反应混合物在减压下浓缩,通过闪蒸色谱法(meoh:ch2cl
2-7.5:
92.5)纯化,得到s68(0.038g,96%)。向s68(0.038g,0.133mmol,1当量)在dmf(1.73ml)的溶液中加入碳酸钾(0.096g,0.69mmol,5当量)。将得到的反应混合物加热到90℃,搅拌7天,或直到通过1hnmr监测反应完成。随后,对反应混合物进行过滤,减压浓缩,用闪蒸色谱法(meoh:ch2cl
2-10:90)纯化粗产物,得到白色固体68(0.027g,71%)。核苷类似物68的数据:[α]
d20
=+16.9(c 1.0in meoh);ir(neat):υ=3261,2988,1686,1272,1203,1047,799cm-1
;1h nmr(600mhz,cd3cn):δ9.43(br s,1h),7.31(d,j=1.1hz,1h),5.48(s,1h),4.27(s,1h),4.15(s,1h),4.03(d,j=8.0hz,1h),3.93(d,j=8.0hz,1h),3.16(s,1h),1.85(d,j=1.1hz,3h);
13
c nmr(150mhz,cd3cn):δ165.1,151.4,135.6,111.0,88.6,80.9,80.3,80.2,75.8,75.2,75.1,13.0.hrms(ei
+
)calcd for c
12h13
n2o5[m+h]
+
265.0819;found 265.0813常规步骤f(α-氟化/羟醛缩合与环己酮/硫吡喃酮35的反应)在-10℃,将醛样品(1.0当量)加入到nfsi(1.0当量),l-脯氨酸(1.0当量),和碳酸氢钠(1.0当量)在dmf(0.75m)的搅拌悬浮液中。通过核磁共振分析当完全转换为α-氟醛时,加入环己酮或硫吡喃酮35(5.0-10.0当量),并将所得到的混合物逐渐加热至室温。共18小时后,用et2o稀释反应混合物,有机层用水洗涤两次,再用盐水洗涤一次。然后将有机层用硫酸镁进行干燥,减压浓缩,粗产物通过闪蒸色谱法纯化。顺式-氟醇68a和反式-氟醇68b的制备按照常规步骤f,在室温下,醛(2.00g,5.86mmol,1.0当量),nfsi(1.85g,5.86mmol,1.0当量),l-脯氨酸(0.674g,5.86mmol,1.0当量)和碳酸氢钠(0.984g,11.71mmol,2当量)在dmf(10ml)的溶液中搅拌2小时。然后加入环己酮(1.15g,11.71mmol),并搅拌反应混合物18小时。然后用乙酸乙酯(100ml)和水(30ml)稀释反应混合物。有机层用盐水(2
×
30ml)洗涤,并用硫酸镁干燥,过滤,减压浓缩。用闪蒸色谱法(25-75%乙酸乙酯在己烷中)纯化粗氟醇68,得到白色固体顺式-氟醇68a(0.92g,产率36%)和反式-氟醇68b(1.21g,产率47%)。顺式-氟醇68a的数据:1h nmr(500mhz,cdcl3):δ8.73(s,1h),8.27(s,1h),7.02(dd,j=50.0,5.6hz,1h),5.82(d,j=6.9hz,1h),4.47(m,1h),2.43(m,1h),2.24(m,1h),2.16(m,1h),2.05(m,1h),1.80
–
1.86(m,2h),1.73(m,1h),1.55
–
1.60(m,2h);
13
c nmr(125mhz,cdcl3):δ209.9,151.5,151.3,151.0,134.0,116.6,92.5(d,j=205.2hz),69.7(d,j=24.4hz),55.3,51.5,51.5,41.5,29.2,26.3,23.5;
19
f nmr(470mhz,cdcl3):δ
–
147.6.反式-氟醇68b的数据:1h nmr(500mhz,cdcl3):δ8.75(s,1h),8.34(s,1h),7.05(dd,j=47.6,7.3hz,1h),5.59(d,j=6.7hz,1h),4.55(m,1h),2.70(m,1h),2.39(m,1h),2.27(m,1h),1.87
–
1.99(m,2h),1.84(m,1h),1.56
–
1.76(m,3h);
13
c nmr(125mhz,cdcl3):210.1,151.6,151.4,151.3,133.8,116.6,91.5(d,j=204.6hz),68.9(d,j=30.5hz),55.2,51.1,41.7,29.1,26.4,23.5顺式-氟醇68a的相对立体化学的测定氟醇68a被转化为核苷86。核苷86的noe分析证实了氟醇68a的相对立体化学。氟醇68a的对映体过量的测定使用1:1的l-:d-脯氨酸的混合物,制备氟醇68a的外消旋品。使用daicel oj-3通过手性sfc分离对映体氟醇;2900psi co2,40℃,3ml/min,梯度为20-30%的25mm异丁胺在异丙醇:co2中持续7分钟,保留时间=2.57分钟和2.77分钟。用相同的方法(94%ee)测定光学富集的氟醇68a的对映体过量。氟醇68b的对映体过量的测定使用1:1的l-:d-脯氨酸的混合物,制备氟醇68b的外消旋品。使用daicel oj-3通过手性sfc分离对映体氟醇;2900psi co2,40℃,3ml/min,梯度为1-20%的25mm二乙胺在甲醇:co2中持续5分钟,保留时间=3.10min和3.32min。用相同的方法(93%ee)测定光学富集的氟醇68b的对映体过量。羟醛缩合加合物69的制备按照常规步骤f,在4℃,将邻苯二甲酰亚胺乙醛(0.050克,0.265毫摩尔)、nfsi(0.84克,0.265毫摩尔)、l-脯氨酸(0.031克,0.265毫摩尔)和2,6-二甲基吡啶(0.031毫升,0.265毫摩尔)的溶液在dmf(0.35毫升)中搅拌15小时。然后加入硫吡喃酮35(0.307克,2.65毫摩尔)并将反应混合物搅拌18小时。通过对粗品的1hnmr光谱分析,确定非对映体的比例为5:1。通过闪蒸色谱法(戊烷:etoac-60:40)纯化,得到白色固体的顺式-和反式-氟醇69的不可分离的混合物(0.075克,产量87%,d.r.=5:1)。氟醇69的数据:1h nmr(600mhz,cdcl3):δ7.93,7.92,7.79,7.79,6.26,6.11,5.37,4.78,3.44,3.25,3.24,3.16,3.11,3.09,3.03,2.99,2.98,2.85,2.80,2.79;
13
c nmr(150mhz,cdcl3):δ212.8,210.2,167.1,167.1,135.1,134.9,131.6,131.5,124.3,124.2,89.6,88.3,70.1,66.1,54.6,53.6,45.7,44.9,34.6,31.3,30.7,30.1;
19
f nmr(470mhz,cdcl3):δ
–
155.5,
–
158.5hrms(ei
+
)calcd for[c
15h14
fno4s+nh4]
+
341.0966;observed 341.0938
羟醛缩合加合物70的制备按照常规步骤f,在4℃,将邻苯二甲酰亚胺乙醛(0.050克,0.265毫摩尔)、nfsi(0.84克,0.265毫摩尔)、l-脯氨酸(0.031克,0.265毫摩尔)和2,6-二甲基吡啶(0.031毫升,0.265毫摩尔)溶液在dmf(0.35毫升)中搅拌16小时。然后加入环己酮(0.275毫升,2.65毫摩尔),并搅拌反应混合物18小时。通过对粗品的1h nmr光谱分析,确定非对映体的比例为5:1。通过闪蒸色谱法(戊烷:etoac-60:40)提纯,得到白色固体的顺式-和反式-氟醇70的不可分离的混合物(0.068克,产量84%,d.r.=5:1)。氟醇70的数据:1h nmr(600mhz,cdcl3):δ7.92,7.91,7.78,7.78,6.29,6.07,5.37,4.63,3.51,2.93,2.92,2.89,2.80,2.44,2.41,2.30,2.25,2.16,2.01.1.99,1.87,1.78,1.71;
13
c nmr(150mhz,cdcl3):δ215.9,213.5,167.1,167.1,134.9,134.8,131.7,131.6,124.1,124.1,89.9,88.3,69.9,65.5,51.8,51.0,43.3,42.7,32.4,28.3,27.8,26.1,25.4,24.8;
19
f nmr(470mhz,cdcl3):δ
–
156.0,
–
160.7hrms(ei
+
)calcd for[c
16h17
fno4]
+
306.1136;observed 306.1135核苷类似物86的制备在0℃下,向68a(100毫克,0.228毫摩尔)在mecn(2.0毫升)中的悬浮液中加入乙酸(131微升,2.285毫摩尔),随后加入三乙酰氧基硼氢化钠(242毫克,1.142毫摩尔)。得到的混合物在室温下搅拌16小时,此时lcms显示以约2.5:1的选择性完全转化为还原产物。然后用水和乙酸乙酯稀释反应混合物。有机层用盐水洗涤,然后用mgso4干燥,过滤,并在减压下浓缩。然后用mecn(2.0ml)稀释粗制的还原产物,并加入氯化铟(50.5mg,0.228mmol)。将得到的反应混合物在50℃下搅拌过夜。然后减压浓缩反应混合物,并通过闪蒸色谱法(25-100%乙酸乙酯在己烷中)提纯,得到白色固体核苷86(43毫克,45%)。核苷类似物86的数据:[α]
d20
=-15.0(c 0.17in meoh);ir(neat):υ=3298,2938,2852,1537,1442,1204,1108cm-1
;1h nmr(600mhz,cdcl3):δ8.68(s,1h),7.98(s,1h),6.11(s,1h),5.59(d,j=4.7hz,1h),4.23(dd,j=4.7,4.4hz,1h),3.64(ddd,j=11.1,11.1,4.0hz,1h),2.08(m,1h),1.72
–
1.82(4h),1.49(m,1h),1.19
–
1.40(m,3h);
13
c nmr(150mhz,cdcl3):δ151.1,150.7,150.1,133.3,116.5,91.0,80.9,76.1,53.4,47.7,40.8,24.8,23.6,23.3hrms(ei
+
)计算得c
14h16
clin3o
2+
419.9970;found 419.9952.核苷86的相对立体化学测定
对核苷86的2d noesy分析支持了所示的立体化学。核苷类似物87的制备在-15℃下,向氟醇70(0.105克,0.344毫摩尔,1.0当量)在mecn(3.00毫升)中的搅拌溶液中加入三乙酰氧基硼氢化四甲基铵(0.453克,1.72毫摩尔,5.0当量)和乙酸(0.190毫升,3.44毫摩尔,10当量)。然后搅拌所得的混合物16小时。然后用罗谢尔盐的饱和溶液稀释反应混合物,用ch2cl2洗涤三次。分离有机层,用mgso4干燥,过滤,减压浓缩。得到的粗产物s70通过闪蒸色谱法(etoac:戊烷-70:30)提纯,得到白色固体s70(0.076克,72%)。向顺式-二醇-氟醇s70(0.076,0.248mmol,1.0当量)在mecn(2.50ml)中的搅拌溶液中加入incl3(0.014g,0.062mmol,0.25当量),并将反应混合物搅拌24小时。反应混合物用ch2cl2稀释,并用饱和碳酸氢钠溶液洗涤。分离有机层,并用mgso4干燥,过滤,减压浓缩。通过对粗产物的1h nmr光谱分析,确定异构体的比例(α:β)为2.5:1。通过闪蒸色谱法(etoac:戊烷-25:75)提纯粗产物87,得到核苷87(α-异构体),为无色油状物(42.7毫克,60%)。核苷类似物87(α-异构体)的数据:[α]
d20
=+46.6(c 0.38in ch2cl2);ir(neat):υ=3475,2935,1708,1370,720cm-1
;1h nmr(600mhz,cdcl3):δ7.88(m,2h),7.77(m,2h),6.13(d,j=5.0hz,1h),4.40(ddd,j=11.8,5.0,4.8hz,1h),4.03(ddd,j=10.6,10.6,4.1hz,1h),3.13(d,j=11.9hz,1h),2.22(m,1h),1.94(m,1h),1.85(m,2h),1.62(dddd,j=11.9,11.9,4.6,3.2hz,1h),1.51(m,1h),1.23
–
1.40(3h);
13
c nmr(150mhz,cdcl3):δ169.1,134.6,132.1,123.8,84.4,81.1,75.3,51.4,31.7,25.4,24.0,24.0hrms(ei
+
)calcd for c
16h18
no4[m+h
+
]288.1230;found 288.1246核苷87的相对立体化学的测定核苷87(α-异构体)的2d noesy分析支持所示的立体化学。核苷类似物88的制备在-15℃下,向氟醇69(0.097克,0.30毫摩尔,1.0当量)在mecn(3.00毫升)中的搅拌溶液中加入三乙酰氧基硼氢化四甲基铵(0.395克,1.50毫摩尔,5.0当量)和乙酸(0.172毫升,1.50毫摩尔,10当量)。所得混合物搅拌16小时。然后用罗谢尔盐的饱和溶液稀释反应
混合物,并用ch2cl2洗涤三次。分离有机层,用mgso4干燥,过滤,并减压浓缩。通过闪蒸色谱法(etoac:戊烷-70:30)提纯粗产物s69,得到白色固体s69(0.068克,70%)。向顺式-二醇-氟醇s69(0.047,0.143毫摩尔,1.0当量)在mecn(1.43毫升)中的搅拌溶液中加入incl3(7.9毫克,0.036毫摩尔,0.25当量),然后将反应混合物搅拌24小时。反应混合物用ch2cl2稀释,并用饱和碳酸氢钠溶液洗涤。分离有机层,用mgso4干燥,过滤,并减压浓缩。通过对粗品的1hnmr光谱分析,确定异构体的比例(α:β)为3:1。通过闪蒸色谱法(etoac:戊烷-40:60)提纯粗产物88,得到88(α-异构体),为无色油(23.7毫克,73%)。核苷类似物88(α-异构体)的数据:[α]
d20
=+18.6(c 2.37in ch2cl2);ir(neat):υ=3475,2923,1774,1709,1373,719cm-1
;1h nmr(600mhz,cdcl3):δ7.88(m,2h),7.77(m,2h),6.13(d,j=4.9hz,1h),4.40(ddd,j=11.5,4.7,4.7hz,1h),4.03(ddd,j=11.2,11.2,3.6hz,1h),3.35(d,j=11.9hz,1h),2.98(dd,j=13.111.9hz,1h),2.82(m,2h),2.69(m,1h),2.50(m,1h),2.10(m,1h),1.74(m,1h);
13
c nmr(150mhz,cdcl3):δ169.2,134.8,131.9,124.0,83.0,80.2,75.2,51.3,33.5,27.6,27.4.hrms(ei
+
)calcd for c
15h19
n2o4s[m+nh
4+
]323.1060;found 323.1037核苷88的相对立体化学测定核苷88(α-异构体)的2d noesy分析支持了所示的立体化学。基于j的构型分析(jbca)使用基于核磁共振j的构型分析对下列化合物的氟立体构型进行赋值,然后使用密度泛函理论计算验证赋值。其他立体中心是基于合成而已知的。核磁共振波谱学通过将几毫克溶解在0.75毫升的dmso-d6中制备nmr样品。然后将这些溶液转移到5毫米的nmr管中。质子化学位移参考2.50ppm的残留dmso-d5,碳化学位移参考39.52ppm的dmso-d6。nmr光谱在配备有5mm三重共振(hcn)氦冷冻探针的600mhz bruker avance iii hd光谱仪或配备有5mm反向prodigy探针的500mhz bruker-avance iii hd谱仪上采集。使
用mnova 12.0.4版本处理数据。获取所有化合物的1h、
13
c、cosy、hsqc和hmbc数据,以分配质子和碳化学位移。使用200ms混合时间采集noesy或roesy光谱,以帮助立体化学测定。dft计算对核磁共振参数、化学位移(d,ppm)和耦合常数(j,hz)的密度泛函理论(dft)进行计算,以验证峰值分配和相对立体构型。首先,使用opls3e力场的混合扭转/低模采样搜索生成一致性的集合,如宏观模型(52)中所实现的。然后,使用gaussian'16(53)中的b3lyp/6-31g(d)模型化学,进一步对小于5kcal/mol的构象体进行dft几何优化和频率测定(以验证势能最小值)。然后,使用wp04/cc-pvdz或wb97x-d/6-31g(d,p)规范(包括质子和碳的原子轨道(giao)方法),从优化的几何结构开始计算各向同性磁屏蔽值s,并根据极化连续体模型(pcm)进行隐式溶剂校正。应用线性比例因子[d=截距-s/-斜率]将s值转换为化学位移,d,单位为ppm。比例因子以前是由rablen等人(54)和lodewyk等人(55)策划的已知结构的大型测试集确定的(1h:截距=31.8465,斜率=-0.9976;
13
c:截距=198.1218,斜率=-0.9816)。使用b3lyp/6-31g(d)模型化学计算耦合常数。使用m06-2x/6-31+g(d,p)和smd溶剂化模型计算吉布斯自由能,并根据玻尔兹曼能量分布对化学位移和耦合常数进行加权。单晶x射线衍射将合适的晶体悬浮在对甲酮(paratone)油中,安装在mitegen micro mount上,然后转移到x射线衍射仪上,使用oxford cryosystems cryostream将其设置为150k。在150k下,在bruker smart仪器上收集数据,该仪器配备了固定在距晶体5.0cm处的apexii ccd区域探测器和1.5kw(45kv,0.65ma)下运行的cukα精细聚焦密封管并用石墨单色仪过滤。使用bruker saint软件包收集和整合数据,并使用多扫描技术(sadabs)校正吸收效应(56)。使用直接方法(sir92)求解结构,并使用shelxl(57)和shelxle(58)进行后续细化。碳原子上的氢原子包含在几何理想化的位置(c
–
h键距离),且未被细化。氢原子的各向同性热参数固定为前一个碳原子的1.2倍。使用mercury(59)和pov-ray(60)绘制图表。表1显示了xrd分析总结。表1:xrd分析总结
大规模制备αfar产品的实例在大规模合成中,没有对反应条件进行额外的优化,在大多数情况下,最终质量中只包括选定的色谱级分。55的大规模制备三个反应并行进行。在15-25℃下向大型反应器中加入dmf(2.1l)和尿嘧啶(300.0g,2.68mol,1.0当量)。然后,在反应器中分别加入dbu(807毫升,5.35摩尔,2.0当量)
和2-溴-1,1-二乙氧基乙烷(483毫升,3.21摩尔,1.2当量)。将反应混合物加热到90℃-100℃,并持续16小时。将反应混合物冷却到25℃,将三批混合物合并并浓缩至干,得到残留物。向残留物中加入水(2.5升),用1mhcl将所得混合物的ph值调节至6-7,并用etoac(2.0升
×
8)萃取。合并的有机层用na2so4干燥,过滤,滤液在减压下浓缩至干,得到残留物。用mbte(3l)在20℃下研磨粗残留物60分钟。粗残留物通过硅胶色谱法(石油醚:etoac:ch2cl2=10:2:1)纯化。烷基化尿嘧啶产品(738克,3.23摩尔,40.3%产率)被分离为白色固体。在15-25℃下,向大型反应器中加入hcl(1m,2.89l,1.0当量)和烷基化胸腺嘧啶产品(660g,2.89mol,1.0当量)。反应混合物被加热到90~100℃并搅拌3小时。在起始材料完全消耗后,将反应混合物冷却到0℃并搅拌30分钟。得到的悬浮液被过滤、干燥,粗产品在下一步使用,无需进一步纯化。得到的醛/水合物(425克,2.76摩尔,95.4%)是一种淡白色的固体。向大型反应器中加入dmf(2800毫升)和醛(400克,2.60摩尔,1.0当量),所得混合物冷却至4℃。然后,在反应器中分别加入nfsi(818克,2.60摩尔,1.0当量)、nahco3(218克,2.60摩尔,1.0当量)和l-脯氨酸(299克,2.60摩尔,1.0当量)。然后反应混合物在4℃下搅拌18小时。hplc(et24077-13-p1a)显示起始材料(rt=0.34)被完全消耗。在4℃下,向反应混合物中滴加二氧环己酮(226克,1.74摩尔,0.67当量)在ch2cl2(1.3升)中的溶液。反应混合物在15~25℃搅拌18小时。hplc(et24077-13-p1a)显示起始材料(rt=1.72分钟)α-氟水合物被完全消耗。向反应混合物中加入14.0升h2o,并用etoac(3.0l
×
8)提取。有机相用na2so4干燥,然后过滤,滤液在减压下浓缩至干,得到一个残留物。残留物通过闪蒸硅胶色谱法(用0~50%的乙酸乙酯/石油醚梯度作为洗脱剂)纯化,得到黄色油状物55(380克,72%产率,d.r.1)。a3的大规模制备在15-25℃下,向大型反应器中加入dmf(1.7l)和胸腺嘧啶(85.0g,0.674mol,1.0当量)。然后,在反应器中分别加入dbu(203毫升,1.35摩尔,2.0当量)和2-溴-1,1-二乙氧基乙烷(122毫升,0.809摩尔,1.2当量)。反应混合物被加热至90℃,并持续14.5小时。将反应混合物浓缩至干,得到一个残留物。向残留物中加入etoac(1.7l)和水(1.7l),分离有机层,水相用etoac(1.7l
×
2)提取。合并的有机相用盐水(500毫升)洗涤,用na2so4干燥,过滤,滤液减压浓缩至干,得到残留物。残留物通过闪蒸硅胶色谱法(5000gsilica闪蒸柱,洗脱剂为30~60%乙酸乙酯/石油醚梯度,@800毫升/分钟)纯化。得到的烷基化胸腺嘧啶产品(80.0克,301毫摩尔,产率22.4%,纯度91.3%)是一种淡白色固体。在15-25℃下,向一个大型反应器中加入hcl(1m,330ml,1.0当量)和烷基化胸腺嘧啶产品(80.0g,0.330mol,1.0当量)。将反应混合物加热到90~100℃,并搅拌15小时。hplc(et17680-15-p1a)显示起始材料(rt=2.77)被完全消耗。混合物浓缩至干,粗品用于下一
步骤,无需进一步纯化。得到的醛/水合物(63.0克混合物)是一种淡白色固体。向一个大型反应器中加入dmf(190毫升)和醛(0.131摩尔,1.0当量),然后将所得混合物冷却到4℃。然后,在反应器中分别加入nfsi(41.3克,0.131摩尔,1.0当量)、nahco3(11.0克,0.131摩尔,1.0当量)和l-脯氨酸(15.1克,0.131摩尔,1.0当量)。反应混合物在4℃搅拌18.5小时。hplc(et17918-3-p1a)显示起始材料(rt=1.99)被完全消耗。在4℃下,向反应混合物中滴加二氧环己酮(11.4克,0.088摩尔,0.67当量)在ch2cl2(200毫升)中的溶液。然后将反应混合物在15~25℃搅拌20.5小时。向混合物中加入570毫升ch2cl2,用水(190毫升
×
3)洗涤有机相。有机相用na2so4干燥,然后过滤,滤液在减压下浓缩至干,得到残留物。残余物通过闪蒸硅胶色谱法(330gsilica闪蒸柱,洗脱剂为0~100%乙酸乙酯/石油醚梯度,@200毫升/分钟)纯化,得到黄色油状物a3(21.0克,76%产率,d.r.3:1(顺:反))。59的16g规模制备将39.0克a3溶解在240毫升的乙酸乙酯中,并通过prep-hlpc再纯化,得到18.0克产物。然后将18.0克产物溶解在240毫升ch2cl2中并减压浓缩,得到17.5克59。最后将17.5克59冷冻干燥,得到15.8克59,为白色固体(纯度94.3%)。顺式-氟醇59的数据:[α]
d20
=-89.4(c 1.1in meoh);ir(neat):υ=2993,1694,1450,1369,1082,1045cm-1
;1hnmr(400mhz,cdcl3):δ8.30(br s,1h),7.57(dd,j=1.3,1.2hz,1h),6.66(ddd,j=42.7,2.3,1.3hz,1h),4.40(dd,j=8.9,1.4hz,1h),4.33(dd,j=17.7,1.4hz,1h),4.12(d,j=17.7hz,1h),4.10(ddd,j=15.4,3.1,2.3hz,1h),3.64(d,j=3.0hz,1h),1.95(d,j=1.2hz,3h),1.52(s,3h),1.46(s,3h);
13
c nmr(100mhz,cdcl3):δ211.2,163.2,149.9,137.1(d,j=4.0hz),111.0,102.1,90.2(d,j=207.8hz),71.6(d,j=2.3hz),70.9(d,j=23.4hz),66.5,23.8,23.4,12.6;
19
f nmr(470mhz,cdcl3):δ
–
177.8hrms(ei
+
)calcd for c
13h18
fn2o6[m+h]
+
317.2929;found 317.1142a5的大规模制备该反应在没有进一步优化的情况下进行。通过柱色谱法纯化粗产物a5,得到16.5克a5(不纯的级分被丢弃)。a6的大规模制备
反应在没有进一步优化的情况下进行。反应仅在16小时后停止。通过prep-hplc纯化粗产物a6,得到36.6克a6(不纯的级分被丢弃)。a8的大规模制备该反应在没有进一步优化的情况下进行。通过prep-hplc纯化粗产物a8,得到47克a8(不纯的级分被丢弃)。一个短的从头合成na的研究进展。本发明研究了-吡唑醛15的-氟化(33)(图2b),发现在dmf(34)中l-脯氨酸和n-氟苯磺酰胺(nfsi)的组合提供了α-氟水合物作为唯一产物(表2)。表2:对α-吡唑醛的αfar进行优化吡唑醛的αfar进行优化
a 1.5当量。b加入的溶剂体积为第1步中的1.25
×
dmf体积。c在第一步中加入的溶剂体积为9
×
dmf体积。在反应混合物中直接加入二氧环己酮8的mecn,得到了产率和对映选择性都很好
的氟醇16a和16b(图2b,条目2)。如图所示,氟醇16a和16b在假异聚碳(用*表示)处以~1.4:1的差向异构体混合物形成,在反应或分离/纯化条件下不相互连接。氟醇16a和16b的还原提供了1,3-顺二醇的混合物,然后用几种路易斯酸中的一种处理该混合物,以促进氟化物通过远端醇官能团的置换,并且实现了使用亲氟sc(otf)3(36)的afd反应,以38%的产率提供作为单β-异构体的na 17(图2b,条目4)。此外,本发明发现用碱(naoh)处理二醇12a和12b的混合物会形成组成随反应时间和碱当量而变化的α-和β-异头nas的混合物(图2b,条目5和6)。使用大量过量的naoh(10当量,条目6),以优异的产率(76%)形成作为唯一产物的β-异构体17。为了进一步研究循环机制,通过快速柱色谱法分离中间体二醇18a和18b,并通过基于j的构型分析和/或衍生物的x射线分析指定它们的相对立体化学。将纯化的顺式-氟醇18a进行afd反应(naoh,ch3cn,图2c),通过sn2过程促进了干净的环化,得到了β-异构体17。类似地,反式-氟醇18b也是通过立体化学转化,环化得到了α-异构体19。在这些相同的反应条件下,α-异构体19差向异构化得到自然构型的β-异构体17,因此两个氟醇羟醛产物可以一起转化为一个自然构型的β-d-na。na 17的对映体纯度(e.r.=95:5,图2b,条目6)表示差向异构体氟尿嘧啶far产品16的对映异构体纯度的平均值。使用αfar和afd策略制备nas。本发明通过几个杂环与溴乙醛二乙缩醛的烷基化制备了一系列乙醛衍生物(图3a)。使用选择氟化物(selectfluor)或nfsi作为亲电氟化剂(f
+
),所得醛类21与二氧环己烷8进行脯氨酸催化的αfar,以提供一系列用杂环尿嘧啶、胸腺嘧啶、三唑、脱氮嗪、吡唑、邻苯二甲酰亚胺、腺嘌呤、2,6-二氯嘧啶或四唑之一的官能化的氟羟醛缩合产物22。这些氟醇的产量和对映体的纯度一般都很高,甚至很好。表3显示了α-(1,2,3)-三唑醛的αfar的优化。表3:α-(1,2,3)-三唑醛的αfar优化三唑醛的αfar优化a1.5当量。b加入的溶剂体积为第一步中的1.25
×
dmf体积。c在第一步中加入的溶剂体积为9
×
dmf体积。在含腺嘌呤的氟醇的情况下,αfar中的竞争(非脯氨酸)催化降低了对映体纯度。每个αfar产物在氟甲基中心分离为差向异构体的混合物,随后进行1,3-同步选择性羰基还原,并通过碱(naoh,图3b)或路易斯酸(图3c)促进afd,如图所示。几个杂环与该过程兼容(图3b-e),尿嘧啶、胸腺嘧啶或腺嘌呤取代的乙醛可以在内源性核糖核苷尿苷(u:24)、5-甲基尿苷(m5u:25)和腺苷(a:31)的简短(共4步)从头合成中得到利用。在这些研究中,促进afd反应的路易斯酸是incl3或sc(otf)3,而吡唑和尿嘧啶衍生的氟醇使用naoh进行环化。在本研究中,除了三唑28、三氟甲基尿嘧啶29和脱氮腺嘌呤32和33之外,nas的产生是单个前体氟醇差向异构体22的对映体纯度的近似平均值。因此,大多数na在afd之后进行差向异构化,提供了将差向异构羟醛产物混合物转化为单一的、天然配置的β-d-核苷类似物的直接方法。对于三氟甲基尿嘧啶29和脱氮腺嘌呤32和33,αfar产物(例如22)被还原、分离,并分别用sc(otf)3或incl3处理。如图3c所示,对于三氟甲基尿嘧啶,只有反式-氟醇经历afd形成29,其在反应条件下没有差向异构化。在脱氮腺嘌呤的情况下,对顺式-氟醇和反式-氟醇进行afd,以分别提供β-和α-异构体32和33,证实了这些反应是通过直接的氟化物置换进行的。一些αfars在大于10g的规模上得到了证明(如25、28、29、30和32(图3c)),本发明注意到当反应在更大的规模上进行时,非对映选择性得到了改善。本发明还发现,可以使用从二氯嘧啶开始的这一系列的反应来制备c-连接的na27,进一步将这一策略的效用扩展到另外一类重要的na。(37)在这里,αfar的主要产物是反式-氟醇,它立体地环化为α-d-核苷类似物,并在反应条件下经历第二次环化事件,形成三环27。除了天然构型的na,通过在αfar中使用d-脯氨酸,这一策略可以很容易地适用于对映体(l-构型)核苷和na的合成(图3e)。因此,l-尿苷(ent-24)和l-构型的naent-28是以这种简单的方式获得的。虽然粗反应混合物通常要用水酸处理,以去除丙酮化物保护基,并能分离出目标na,但取消这一步骤后,本发明可以直接分离出c3'/c5'保护的na(例如,34和35,图3d)。为了证明这些受丙酮保护的na可以用标准方案进一步衍生化,本发明制备了几种c2'修饰的na,包括c2'-氧(36)、c2'-脱氧(37)、c2'-3
°
醇(38)和c2'-epi(39)(图3f)。表4和表5中显示了afd反应的优化情况。表4:afd反应的优化
36。因此,在这种情况下,镁醇盐42a和42b中的每一个都选择性地使用互补的碱或路易斯酸促进的afd工艺进行循环,以获得α-l和α-d构型的na。本发明还检查了几种其他有机镁试剂与含有三唑、脱氮腺嘌呤、胸腺嘧啶、吡唑或三氟甲基尿嘧啶功能的氟醇羟醛缩合加合物的反应(图4a)。在这项研究中,本发明发现1,2-加成反应中的立体选择性程度取决于溶剂和杂环。例如,将memgbr添加到thf中的酮氟醇中,得到了与在ch2cl2中产生的成分不同的叔醇的混合物。将memgbr添加到被三唑取代的酮氟醇中主要得到1,3-顺-二醇,其经历afd以产生自然配置的naα-d-48。因此,一系列脱氮腺嘌呤取代的na35-39很容易以α-和β-异构体的形式被获取。在这些研究中,碱促进的afd导致了c3',c5'-保护的na(如49-54),而路易斯酸促进的afd导致了脱保护或保护基迁移(如44,47和48)。正如图4所总结的,一系列密集功能化的c4'-修饰的na可以从相应的酮氟醇羟醛缩合加合物中快速获得,包括被甲基、环丙基、芳基和炔基取代的na。每个c4'-甲基、环丙基、对甲氧基苯基、对氯苯基、炔基na 43-54的制备总共只需要3或4个步骤。1,2-加成反应的优化情况见表6。表6:1,2-加成反应的优化加成反应的优化a3当量。b0.10 m。c通过1h nmr分析粗制的反应混合物来确定。d独立的产量。用于合成uprifosbuvir的大规模αfar。本发明研究了从900克尿嘧啶开始的d-尿苷衍生物56的合成。在没有任何额外优化的情况下,本发明能够生成约380克的羟醛缩合加合物55(图2b),它可以通过碱促afd转化为受保护的尿苷56,产量很高。c2'-oh官能团的氧化,然后在四氢呋喃中脱保护并加入memgbr,得到叔醇57。这个后来的化合物是以前报道的大规模生产mk-3682(uprifosbuvir:58)的一个中间体(38)。亚氨基核苷、脱氧核苷和锁定核酸的合成。本发明还评估了该方法在获得一类不寻常的nas(称为亚氨基核苷或4'-氮杂核苷)时的效用,其中呋喃氧被氮原子取代。因此,在一个实施例(图4c)中,显示了使用苄胺对
氟醇-羟醛加合物59(如图所示作为单一非对映体分离)进行还原胺化,然后进行基本处理,直接以良好的产率得到β-d构型的亚氨基核苷60。为了证明这条路线对获得c2'和c4'修饰的na的效用,本发明制备了一个c4'修饰的、c2'-脱氧na(图4d)。在这里,c4'-烯丙基胸腺嘧啶61通过将烯丙基溴化镁加入到氟醇羟醛缩合加合物59中,然后进行碱促进的afd,以良好的产率制备。然后通过barton-mccombie脱氧反应,仅用6个步骤就从胸腺嘧啶中得到了4'-烯丙基na 62。为了证明这一过程对na合成的实用性,本发明研究了c4'功能化制备锁定核酸(lnas)的方法。为了实现统一的lna合成,本发明评估了烷基溴化镁对含胸腺的羟醛缩合加合物59的加成,发现该反应得到了两个非对映体加成产物63和64,总产率很高。主要产物通过与naoh反应直接转化为不寻常的lna67,naoh促进了afd反应和随后在游离醇功能和炔之间的环化,总产率非常好。这个4步的总合成与报道的类似的尿嘧啶lna67(40)的23步路线相比较好。我们还能够通过简单地对1,2-加成产物64进行afd,产生不寻常的烷基功能化lna68,这是一个以前没有报道过的核苷化学的支架。从这里,形成2,2'-脱水胸苷,然后在温暖的dmf(41)中进行脱保护和碱处理,得到lna68。这个独特的支架为通过标准的点击反应或sonagashira偶联反应实现进一步的多样化奠定了基础。参考文献1.g.m.blackburn,gait,m.j.,loakes,d.,williams,d.m.,ed.,nucleic acids in chemistry and biology,(royal society of chemistry,cambridge,uk,2006),pp.503.2.c.m.galmarini,j.r.mackey,c.dumontet.nucleoside analogues and nucleobases in cancer treatment.lancet oncol.3,415-424(2002).3.e.de clercq.highlights in antiviral drug research:antivirals at the horizon.med.res.rev.33,1215-1248(2013).4.l.p.jordheim,d.durantel,f.zoulim,c.dumontet.advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases.nat.rev.drug discov.12,447-464(2013).5.d.m.huryn,m.okabe.aids-driven nucleoside chemistry.chem.rev.92,1745-1768(1992).6.j.shelton et al.metabolism,biochemical actions,and chemical synthesis of anticancer nucleosides,nucleotides,and base analogs.chem.rev.116,14379-14455(2016).7.b.ewald,d.sampath,w.plunkett.nucleoside analogs:molecular mechanisms signaling cell death.oncogene 27,6522-6537(2008).8.k.l.seley-radtke,m.k.yates.the evolution of nucleoside analogue antivirals:a review for chemists and non-chemists.part 1:early structural modifications to the nucleoside scaffold.antiviral res.154,66-86(2018).9.m.k.yates,k.l.seley-radtke.the evolution of antiviral nucleoside analogues:a review for chemists and non-chemists.part ii:complex modifications to the nucleoside scaffold.antiviral res.162,5-21(2019).
goals.chem.soc.rev.40,5680-5689(2011).40.p.p.seth,e.e.swayze.(2008).6-disubstituted or unsaturated bicyclic nucleic acid analogs.us 8278283 b2.ionis pharmaceuticals.41.t.yamaguchi,m.horiba,s.obika.synthesis and properties of 2
′‑
o,4
′‑
c-spirocyclopropylene bridged nucleic acid(scpbna),an analogue of 2
′
,4
′‑
bna/lna bearing a cyclopropane ring.chem.commun.51,9737-9740(2015).42.w.ren et al.revealing the mechanism for covalent inhibition of glycoside hydrolases by carbasugars at an atomic level.nat.commun.9,3243(2018).43.a.quintard,j.rodriguez.bicatalyzed three-component stereoselective decarboxylative fluoro-aldolization for the construction of elongated fluorohydrins.acs catalysis 7,5513-5517(2017).44.t.c.britton,m.e.letourneau.(1995).process for anomerizing nucleosides.us 5,420,266.eli lilly and company.45.a.m.downey,c.richter,r.pohl,r.mahrwald,m.hocek.direct one-pot synthesis of nucleoside from unprotected or 5-o-monoprotected d-ribose.org.lett.17,4604-4607(2015)46.z.-q.xu,y.-l.qui,s.chokekijchai,h.mitsuya,j.zemlicka.unsaturated acyclic analogs of 2
’‑
deoxyadenosine and thymidine containing fluorine:synthesis and biological activity.j.med.chem.38,875(1995)47.e.moyroud,e.biala,p.strazewski.synthesis and enzymatic digestion of an rna nonamer in both enantiomeric forms.tetrahedron 56,1475-1484(2000)48.a.hadj-bouazza,r.zerrouki,p.krausz,g.laumond,a.m.aubertin,y.champavier.new acyclonucleosides:synthesis and anti-hiv activity.nucleosides,nucleotides,and nucleic acids 24,1249-1263,(2005)49.y.mehellou,r.valente,h.mottram,e.walsby,k.i.mills,j.balzarini,c.mcguigan.phosphoramidates of 2
’‑
beta-d-arainouridine(arau)as phosphate prodrugs;design,synthesis,in vitro activity and metabolism.bioorg.med.chem.18,2439-2446,(2010).50.a.f.cook,j.g.moffatt.sulfoxide-carbodiimide reactions.vi.synthesis of 2
’‑
and 3
’‑
ketouridines.j.am.chem.soc.89,2697,(1967)51.s.f.jenkinson,n.a.jones,a.moussa,a.j.stewart,j.heinz,g.w.j.fleet.anomeric stereospecific synthesis of 2
’‑
c-methylβ-nucleosides;the holy reaction of cyanamide with 2-c-methyl-d-arabinose.tetrahedron letters 48,4441-4444,(2007)52.release 2018-3:macromodel,llc,new york,ny(2019).53.gaussian 16,revision c.01,m.j.frisch et al.gaussian,inc.,wallingford ct(2016).
54.p.r.rablen,s.a.pearlman,j.finkbiner.a comparison of density functional methods for the estimation of proton chemical shifts with chemical accuracy.j.phys.chem.a 103,7357-7363(1999)55.m.w.lodewyk,m.r.siebert,d.j.tantillo.computational prediction of 1
h and 13
c chemical shifts:a useful tool for natural product,mechanistic,and synthetic organic chemistry.chem.rev.112,1839-1862(2012)56.bruker,apex3,saint and sadabs,bruker axs inc.,madison,wi(2016).57.g.m.sheldrick.crystal structure refinementwith shelxl.acta crystallogr.sect.c struct.chem.71,3-8(2015).58.c.b.h
ü
bschle,g.m.sheldrick,b.dittrich.shelxle:a qt graphical user interface for shelxl.j.appl.crystallogr.44,1281-1284(2011).59.c.f.macrae,i.j.bruno,j.a.chisholm,p.r.edgington,p.mccabe,e.pidcock,l.rodriguez-monge,r.taylor,j.van de streek and p.a.wood.mercury csd 2.0
–
new features for the visualization and investigation of crystal structures.j.appl.cryst.41,466-470(2008)60.t.d.fenn,d.ringe,g.a.petsko.povscript+:a program for model and data visualization using persistence of vision ray-tracing.j.appl.crystallogr.36,944-947(2003).所有引用均以参考的形式收录于此。本发明已就一个或多个实施例进行了描述。然而,对于本领域的技术人员来说,显然可以在不脱离权利要求书中所定义的本发明的范围的情况下做出一些变化和修改。因此,尽管这里公开了本发明的各种实施例,但根据本领域技术人员的公知常识,可以在本发明的范围内做出许多调整和修改。这种修改包括用已知的等价物替代本发明的任何方面,以便以基本相同的方式达到相同的结果。数字范围包括定义该范围的数字。在说明书中,“包括”一词是作为一个开放性术语使用的,基本上等同于“包括但不限于”这一短语,而“包括”一词具有相应的含义。然而,应该理解的是,在这里使用“包括”或“包含”或具有相同词根的变体时,也可以考虑将其变体或修改为“由”,即排除未指定的任何元素、步骤或成分,或“基本上由”或“基本由”,即限定为指定的材料或所提及的步骤,以及那些不会对所要求的发明的基本和新颖特征有实质性影响的材料。此外,除非上下文另有说明,本发明中所有描述的元素的任何排列和组合都应被视为本发明的描述所公开。在此引用参考文献不应解释为承认这些参考文献是本发明的现有技术。所有的出版物都通过引用而被纳入本发明,就像每一个单独的出版物都被具体和单独地指出要通过引用而被纳入本文一样,而且就像在本文中完全阐述的一样。本发明包括基本上如前所述并参照实施例和附图的所有实施方案和变化。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1