一种奥洛他定氘标记代谢物的制备方法与流程
1.本发明涉及一种化合物的制备方法,具体设计一种奥洛他定氘标记代谢物的制备方法。
技术背景
2.奥洛他定(olopatadine),是一种选择性二苯并氧杂环庚三烯-2-乙酸类组胺h1受体拮抗剂及肥大细胞稳定剂,能够抑制速激肽及其它化学递质,如组胺、花生四烯酸、血栓素、白三烯等的释放。临床用于过敏性鼻炎、过敏性结膜炎、荨麻疹、伴有瘙痒症状的皮肤疾病(湿疹、多形性渗出性红斑等)和哮喘。口服治疗过敏性鼻炎、荨麻疹、伴有瘙痒症状的皮肤疾病(湿疹、多形性渗出性红斑等)。1%滴眼液治疗过敏性结膜炎。0.6%鼻喷剂用于缓解6岁及以上患者季节性过敏性鼻炎症状。
3.随着时代的进步、科技水平的提高,人们对药品上市前必须对药品进行质量、安全性和效能科学评价的重要性等有了更加充分的认识,其中与药品质量密切相关的是药物所含氘标记代谢物的控制。氘标记代谢物往往与药品安全性有关,且在少数情况下与效能也有关。因此,控制氘标记代谢物的水平,在药物开发研究过程中越来越受到医药工作者的重视。
4.本发明提供的奥洛他定氘标记代谢物的合成方法,尚未见报道,对奥洛他定氘标记代谢物进行相关的药理学、药动学等研究,氘标记的药物可以用来追踪药物在生物体内的代谢过程,在临床药代动力学研究中具有极大的应用研究价值。
技术实现要素:
5.发明目的:本发明的目的是为了解决现有技术的不足,提供一种工艺设计合理,产率高,操作过程方便可控的新型药物分子奥洛他定氘标记代谢物的合成方法。
6.技术方案:为了实现以上目的,本发明采取的技术方案为:
7.一种奥洛他定氘标记代谢物的制备方法,其特征在于,包括如下步骤:
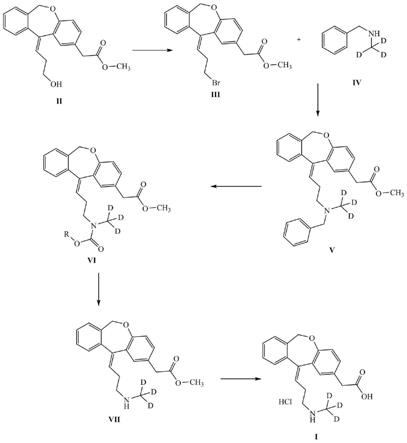
8.(1)取化合物ⅱ在溴化试剂的作用下反应得到化合物ⅲ;
[0009][0010]
(2)取化合物ⅲ与标记的化合物ⅳ在碱作用下于非质子性溶剂中,加热搅拌反应,反应完
[0011]
处理后经柱色谱提纯后得到化合物
ⅴ
;
[0012][0013]
(3)取化合物
ⅴ
溶于有机溶剂中,加入氯甲酸酯,反应得到化合物ⅵ;
[0014][0015]
(4)取化合物ⅵ溶于质子性溶剂中,反应完得到化合物ⅶ;
[0016][0017]
(5)取化合物ⅶ溶于有机溶剂中,在冰浴条件下,加入无机碱,进行水解反应,反应完后调酸析出的固体经干燥后得到化合物ⅰ,即奥洛他定氘标记代谢物
[0018][0019]
本发明制备得到的奥洛他定氘标记代谢物,化学名称为(z)-2-(11-(3-((甲基-d3)氨基)亚丙基)-6,11-二氢二苯并[b,e]奥克西平-2-基)乙酸盐酸盐,分子量为362.87,分子式为c
20h19
d3clno3。
[0020]
作为优选方案,以上所述的奥洛他定氘标记代谢物的制备方法,步骤(1)所述的溴化试剂为三溴化磷、三苯基膦/四溴化碳、氢溴酸等;
[0021]
作为优选方案,以上所述的奥洛他定氘标记代谢物的制备方法,步骤(1)所述化合物ⅱ和溴化试剂的摩尔用量比为1:1~1:3,优选为1:1.5。
[0022]
作为优选方案,以上所述的奥洛他定氘标记代谢物的制备方法,步骤(2)所述的碱为氢氧化钠、氢氧化钾、碳酸钾、碳酸钠、三乙胺等。
[0023]
作为优选方案,以上所述的奥洛他定氘标记代谢物的制备方法,步骤(2)所述的非质子性溶剂为二甲基亚砜、n,n-二甲基甲酰胺、乙腈、四氢呋喃等。
[0024]
作为优选方案,以上所述的奥洛他定氘标记代谢物的制备方法,步骤(2)所述的底物(化合物ⅲ与标记的化合物ⅳ)与碱的摩尔用量比为1:1~1:3,优选为1:1.5。其中化合物ⅲ与标记的化合物ⅳ的摩尔用量比为1:1~1.7。
[0025]
作为优选方案,以上所述的奥洛他定氘标记代谢物的制备方法,步骤(3)所述的有机溶剂为二氯甲烷、1,2-二氯乙烷、甲苯、正己烷等。
[0026]
作为优选方案,以上所述的奥洛他定氘标记代谢物的制备方法,步骤(3)所述的氯甲酸酯为氯甲酸乙酯、氯甲酸-1-氯乙酯、氯甲酸三氯乙酯、氯甲酸甲酯等。
[0027]
作为优选方案,以上所述的奥洛他定氘标记代谢物的制备方法,步骤(4)所述的质子性溶剂为甲醇、乙醇、异丙醇等。
[0028]
作为优选方案,以上所述的奥洛他定氘标记代谢物的制备方法,步骤(5)所述的有机溶剂为甲醇,乙醇,四氢呋喃,二氧六环等,所述的无机碱为氢氧化钠,氢氧化钾,氢氧化锂等。
[0029]
有益效果:本发明提供的奥洛他定氘标记代谢物的制备方法具有以下优点:
[0030]
1.本发明提供的新型药物分子奥洛他定氘标记代谢物的合成方法,工艺设计合理,操作方法简单、原料易得、合成效率高、纯度高、反应过程可控和环境保护效果好。
[0031]
2.并且本发明制备得到的奥洛他定氘标记代谢物,可为奥洛他定的质量、安全性和效能科学评价提供重要依据,具有重要的应用价值。
附图说明
[0032]
图1为本发明提供的奥洛他定氘标记代谢物的制备工艺流程图。
[0033]
图2为本发明化合物ⅰ的1h-nmr图(dmso-d6)。
[0034]
图3为本发明的化合物ⅰ的1h-nmr图(dmso-d6+d2o)。
[0035]
图4是本发明的化合物ⅲ的1h-nmr图(dmso-d6)。
具体实施方式
[0036]
以下的实施例在于详细说明本发明,但不是限制本发明。
[0037]
实施例1
[0038]
如图1所示:奥洛他定氘标记代谢物的制备方法,具体包括以下步骤:
[0039]
(1)化合物ⅲ的制备:取化合物ⅱ(12g,37mmol)溶于100毫升氢溴酸中,50摄氏度反应过夜后得到化合物ⅲ(13g),核磁显示正确,收率90.8%,化合物ⅲ的1h nmr(如图4):dmso-d6:δ2.93(m,2h),3.60(s,5h),3.67(t,2h),5.24(br,2h),5.66(t,1h),6.77(d,1h),7.05-7.11(m,2h),7.28-7.30(m,1h),7.31-7.43(m,3h)。
[0040][0041]
(2)化合物
ⅴ
的制备:取化合物ⅲ(13g,33mmol)和化合物ⅳ(4.6g,37mmol)溶于于四氢呋喃(650ml)中,加入7g碳酸钾,回流反应20h后,抽滤浓缩后,经柱色谱提纯(展开剂:
二氯甲烷-甲醇:100:1)后得到化合物
ⅴ
(13.0g),质谱显示正确,ms:m/z,431.2,收率为90.06%。
[0042][0043]
(3)化合物ⅵ的制备:取化合物
ⅴ
(13g,30mmol)溶于甲苯中(130ml)中,加入氯甲酸1-氯乙酯(8.63g,60mmol),60摄氏度反应2h后直接浓缩干燥后得到13.4g黄色油状物ⅵ,质谱显示正确,ms:m/z,447.2,收率为99%。
[0044][0045]
(4)化合物ⅶ的制备:取化合物ⅵ(13.4g,30mmol)溶于乙醇(250ml)中,回流反应过夜后至原料反应完全,反应完浓缩后,用柱色谱分离(展开剂:二氯甲烷-甲醇:20:1)得8.0g淡黄油状物ⅶ,质谱显示正确,ms:m/z,341.2,收率为78.48%。
[0046][0047]
(5)化合物ⅰ的制备:取化合物ⅶ(8.0g,23mmol)溶于四氢呋喃(100ml)中,在室温下,加入2m的氢氧化钠溶液8ml,室温反应2小时,浓缩后溶于水中,用稀盐酸调成酸性后,抽滤,真空干燥后得5.30g类白色固体化合物ⅰ,收率为62.30%。化合物ⅰ的1h nmr的图谱如图2和图3:dmso-d6+d2o:δ2.73(m,2h),3.10(t,2h),3.53(s,2h),4.80-5.60(br,2h),5.66(t,1h),6.81(d,1h),7.03-7.04(s,1h),7.11-7.13(d,1h),7.30-7.45(m,4h)。
[0048][0049]
实施例2
[0050]
工艺流程图如图1所示:奥洛他定氘标记代谢物的制备方法,具体包括以下步骤:
[0051]
(1)化合物ⅲ的制备:取化合物ⅱ(5g,15mmol)溶于50毫升氢溴酸中,100摄氏度反应过夜后得到化合物ⅲ(5.6g),h-nmr数据同实施例1,收率93.9%。
[0052]
(2)化合物
ⅴ
的制备:取化合物ⅲ(13g,33mmol)和化合物ⅳ(4.6g,37mmol)溶于于四氢呋喃(650ml)中,加入2g氢氧化钠,回流反应20h后,抽滤浓缩后,经柱色谱(色谱条件同实施例1)提纯后得到化合物
ⅴ
(13.5g),ms数据同实施例1,收率为93.53%。
[0053]
(3)化合物ⅵ的制备:取化合物
ⅴ
(13g,30mmol)溶于1,2二氯乙烷中(130ml)中,加入氯甲酸乙酯(6.55g,60mmol),60摄氏度反应3h后直接浓缩干燥后得到13.0g黄色油状物ⅵ,ms数据同实施例1,收率为96.3%。
[0054]
(4)化合物ⅶ的制备:取化合物ⅵ(13.4g,30mmol)溶于异丙醇(250ml)中,回流反应过夜后原料反应完全,反应完浓缩后,用柱色谱(色谱条件同实施例1)分离得7.5g淡黄油状物ⅶ,ms数据同实施例1,收率为73.6%。
[0055]
(5)化合物ⅰ的制备:取化合物ⅶ(8.0g,23mmol)溶于甲醇(100ml)中,在室温下,加入2m的氢氧化钠溶液8ml,室温反应2小时后浓缩后溶于水中用稀盐酸调成酸性后,抽滤,真空干燥后得5.5g类白色固体化合物ⅰ,收率为64.66%。化合物ⅰ的1h nmr的图谱如图2和图3。h-nmr数据同实施例1。
[0056]
实施例3
[0057]
工艺流程图如图1所示,奥洛他定氘标记代谢物的制备方法,具体包括以下步骤:
[0058]
(1)化合物ⅲ的制备:取化合物ⅱ(5g,15mmol)溶于50毫升四氢呋喃中,加入三苯基膦(4.85g,18mmol)和四溴化碳(7.67g,23mmol),室温反应过夜后浓缩经柱色谱提纯后得到化合物ⅲ(5.7g),h-nmr数据同实施例1,收率95.6%。
[0059]
(2)化合物
ⅴ
的制备:取化合物ⅲ(13g,33mmol)和化合物ⅳ(6.3g,50mmol)溶于于四氢呋喃(650ml)中,加入2g氢氧化钠,回流反应20h后,抽滤浓缩后,经柱色谱(色谱条件同实施例1)提纯后得到化合物
ⅴ
(14.0g),ms数据同实施例1,收率为97.0%。
[0060]
(3)化合物ⅵ的制备:取化合物
ⅴ
(13g,30mmol)溶于甲苯中(130ml)中,加入氯甲酸三氯乙酯(12.79g,60mmol),20摄氏度反应2h后直接浓缩干燥后得到13.5g黄色油状物ⅵ,ms数据同实施例1,收率为99.9%。
[0061]
(4)化合物ⅶ的制备:取化合物ⅵ(13.4g,30mmol)溶于乙醇(130ml)中,回流反应过夜后原料反应完全,反应完浓缩后,用柱色谱(色谱条件同实施例1)分离得8.2g淡黄油状物ⅶ,ms数据同实施例1,收率为80.4%。
[0062]
(5)化合物ⅰ的制备:取化合物ⅶ(8.0g,23mmol)溶于四氢呋喃(100ml)中,在室温下,加入2m的氢氧化钠溶液8ml,40℃反应1小时后浓缩后溶于水中用稀盐酸调成酸性后,抽滤,真空干燥后得6.0g类白色固体化合物ⅰ,收率为70.5%。化合物ⅰ的1h nmr的图如图2和图3。,h-nmr数据同实施例1。
[0063]
经过比对分析实施例1、实施例2和实施例3可看出,不同的反应原料,反应原料的用量配比和反应条件不同,合成产物的得率并不相同,其中实施例3的各步骤得率均高于实施例1和实施例2,说明按实施例3的反应条件最佳。取得了比较好的技术进步。
[0064]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应
视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1