一种吲哚咔唑类化合物的制备方法
1.本发明涉及有机合成技术领域,尤其涉及一种吲哚咔唑类化合物的制备方法。
背景技术:
2.吲哚咔唑类生物碱是一类非常重要的来源于微生物的代谢产物,其化学结构主要由吡咯烷酮、吲哚咔唑环以及糖基三部分组成。自日本科学家omura等人于1977年首次从海洋微生物streptomyces staurosporeus中分离到staurosporine以来,迄今为止已经陆续有约130多个结构新颖的吲哚咔唑类生物碱被分离得到,并表现出很好的生物活性。其中最具代表性的有staurosporine、rebeccamycin、k252a、tjipanazole f2和tjipanazole d等。
3.吲哚咔唑类生物碱不仅数量众多,结构复杂,而且具有许多重要的药理活性,如抗肿瘤、抗高血压、抗真菌、抗细菌、抗疟疾、抗结核、抗血小板凝集、抑制免疫、保护神经和杀虫等活性,在医学及农业领域有很好的应用前景。目前已经有多个吲哚咔唑类生物碱进入了临床研究,如pkc412(midostaurin)于2008年在美国启动了治疗aml的ⅲ期临床研究,并于2017年获得fda批准上市;ucn-01已经完成了治疗复发性难治急性白血病以及高危性骨髓增生异常综合症等的ⅰ期临床研究,治疗乳腺癌和淋巴瘤等的ⅱ期临床研究,以及治疗转移性黑素瘤的ⅱ期临床研究;cep-701处于治疗aml的ⅲ期临床阶段,且于2006年获得美国fda批准作为治疗aml的孤儿药;cep-2563作为一种受体酪氨酸激酶抑制剂,已经完成了治疗反复性实体瘤的ⅰ期临床研究;cep-1347已经完成对帕金森疾病治疗的ⅱ期临床研究,进入了ⅲ期临床试验;cep-7055具有抗新生血管生长活性以及抗肿瘤等生物活性,处于实体瘤的ⅰ期临床试验之中;nb-506抑制拓扑异构酶i的选择性增强,目前正处于癌症治疗的ⅰ/ⅱ期临床研究之中;edotecarin同样具有高效的抑制拓扑异构酶ⅰ的活性,而且更加的稳定,目前也正处于ⅰ/ⅱ期临床研究之中;bms-250749具有很好的拓扑异构酶ⅰ抑制活性,临床试验中比市售抗癌药物伊立替康(cpt-11)具有更广谱的抗肿瘤活性。
4.但随着研究的深入,人们也发现虽然吲哚咔唑类生物碱的活性显著,但其天然来源非常有限、临床特异性较差等缺点很大程度上制约了其新药的研发进程。因此,发展新的合成方法,制备更多结构新颖、选择性较高的吲哚咔唑类生物碱,对其进行深入的构效关系研究就成为了一种必然趋势。
技术实现要素:
5.本发明的目的在于提供一种吲哚咔唑类化合物的制备方法,能够简洁、高效的制备一系列吲哚咔唑类化合物。
6.为了实现上述发明目的,本发明提供以下技术方案:
7.本发明提供了一种吲哚咔唑类化合物的制备方法,包括以下步骤:
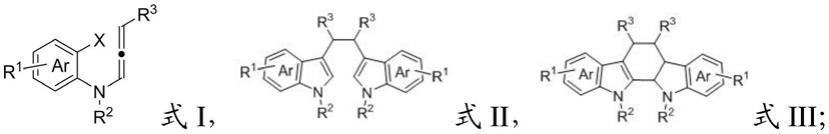
8.将具有式i所示结构的联烯底物、光催化剂和有机碱溶于第一有机溶剂中,在光照条件下进行自由基串联反应,得到具有式ii所示结构的自由基串联反应产物;
9.将所述具有式ii所示结构的自由基串联反应产物和酸溶于第二有机溶剂中,发生
曼尼希环化反应,得到具有式iii所示结构的吲哚咔唑类化合物;
[0010][0011]
所述式i、式ii和式iii中,ar为苯、萘、蒽、菲、芴、吡啶、喹啉、异喹啉、呋喃、苯并呋喃、噻吩、苯并噻吩或苯并噻唑;x为氯、溴或碘;r1为不同个数和取代位置的氢、烷基、烷氧基、烯基、炔基、芳基、卤素、羟基、巯基、硝基、氰基、羧基、酯基、醛基、酰基、酰氧基、胺基、取代胺基或酰胺基;r2和r3独立地为氢、烷基、烯基、炔基、芳基、氰基、酯基、醛基、酰基、酰氧基或酰胺基。
[0012]
优选的,所述光催化剂为金属光敏剂或有机光敏剂。
[0013]
优选的,所述金属光敏剂包括铱催化剂、钌催化剂、钯催化剂或铜催化剂;所述有机光敏剂包括曙红y或玫瑰红。
[0014]
优选的,所述有机碱为三乙胺、三甲胺、二异丙基乙基胺、吡啶、2,6-二甲基吡啶、2,6-二叔丁基吡啶、2,4,6-三甲基吡啶、4-二甲氨基吡啶、1,4-二氮杂二环[2.2.2]辛烷、1,8-二氮杂二环[5.4.0]十一碳-7-烯、四甲基胍、三乙烯二胺、四甲基乙二胺、n-甲基吗啉或n,n,n',n”,n
”‑
五甲基二亚乙基三胺;
[0015]
所述第一有机溶剂为正己烷、环己烷、庚烷、苯、甲苯、二甲苯、三甲苯、三氟甲苯、氯苯、四氯化碳、氯仿、二氯甲烷、1,2-二氯乙烷、四氢呋喃、乙醚、汽油、二硫化碳、氯丙烷、溴乙烷、异丙醚、硝基甲烷、乙酸丁酯、正戊烷、二氧六环、乙酸甲酯、甲基叔丁基醚、石油醚、丙酮、乙腈、甲醇、乙醇、异丙醇、正丙醇、正丁醇、异丁醇、叔丁醇、水、吡啶、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲基亚砜、乙酸乙酯、三氟乙醇或六氟异丙醇。
[0016]
优选的,所述具有式i所示结构的联烯底物和光催化剂的摩尔比为1:(0.001~1),所述具有式i所示结构的联烯底物和有机碱的摩尔比为1:(1~20)。
[0017]
优选的,所述自由基串联反应的温度为-50℃~100℃,光照波长为300nm~700nm,反应时间为10~30h。
[0018]
优选的,所述酸为质子酸或路易斯酸。
[0019]
优选的,所述质子酸为盐酸、硫酸、磷酸、硝酸、硼酸、氟硼酸、苯磺酸、氢氟酸、氢溴酸、氢碘酸、高氯酸、高碘酸、甲酸、乙酸、丙酸、丁二酸、己二酸、庚酸、甲磺酸、三氟甲磺酸、苯甲磺酸、对甲苯磺酸、樟脑磺酸或苯甲酸,所述路易斯酸为氯化锌、三氯化铝、三氟化硼、四氟硼酸锂、三氟甲磺酸银或三氟甲磺酸锌。
[0020]
优选的,所述具有式i所示结构的联烯底物和酸的摩尔比为1:(1~200)。
[0021]
优选的,所述曼尼希环化反应的温度为-20℃~130℃,时间为6~12h。
[0022]
本发明以具有式i所示结构的联烯类化合物为原料,通过光激发发生单电子转移引发自由基串联反应,并在酸性条件下发生曼尼希环化反应,一锅合成吲哚咔唑类化合物,方法简单、高效,原料和试剂便宜易得,反应产率高,副产物少,反应的化学和区域选择性高(光照下原位生成的芳基自由基对联烯的中间碳进行选择性加成后随即发生自由基的二聚);本发明方法操作简单,适于工业化生产和市场推广应用。
具体实施方式
[0023]
本发明提供了一种吲哚咔唑类化合物的制备方法,包括以下步骤:
[0024]
将具有式i所示结构的联烯底物、光催化剂和有机碱溶于第一有机溶剂中,在光照条件下进行自由基串联反应,得到具有式ii所示结构的自由基串联反应产物;
[0025]
将所述具有式ii所示结构的自由基串联反应产物和酸溶于第二有机溶剂中,发生曼尼希环化反应,得到具有式iii所示结构的吲哚咔唑类化合物;
[0026][0027]
所述式i、式ii和式iii中,ar为苯、萘、蒽、菲、芴、吡啶、喹啉、异喹啉、呋喃、苯并呋喃、噻吩、苯并噻吩或苯并噻唑;x为氯、溴或碘;r1为不同个数和取代位置的氢、烷基、烷氧基、烯基、炔基、芳基、卤素、羟基、巯基、硝基、氰基、羧基、酯基、醛基、酰基、酰氧基、胺基、取代胺基或酰胺基;r2和r3独立地为氢、烷基、烯基、炔基、芳基、氰基、酯基、醛基、酰基、酰氧基或酰胺基。
[0028]
在本发明中,未经特殊说明,所用原料均为本领域熟知的市售产品。
[0029]
本发明将具有式i所示结构的联烯底物、光催化剂和有机碱溶于第一有机溶剂中,在光照条件下进行自由基串联反应,得到具有式ii所示结构的自由基串联反应产物,
[0030][0031]
在本发明中,所述式i、式ii和式iii中的苯环代表的不是一个苯环,而是若干芳香环,特此说明。
[0032]
在本发明的实施例中,所述具有式1所示结构的联烯底物为
[0033]
在本发明中,所述光催化剂优选为金属光敏剂或有机光敏剂;所述金属光敏剂优选包括铱催化剂、钌催化剂、钯催化剂或铜催化剂;所述铱催化剂优选包括[ir(dfppy)2(dtbpy)]pf6或fac-[ir(ppy)3];所述钌催化剂优选包括[ru(bpy)3]cl2.6h2o或[ru(bpz)3](pf6)2;所述钯催化剂优选包括pd(oac)2或pd(pph3)4;所述铜催化剂优选包括(dpephos)(bcp)cu]pf6。
[0034]
在本发明中,所述有机光敏剂优选包括曙红y或玫瑰红。
[0035]
在本发明中,所述有机碱优选为三乙胺、三甲胺、二异丙基乙基胺、吡啶、2,6-二甲基吡啶、2,6-二叔丁基吡啶、2,4,6-三甲基吡啶、4-二甲氨基吡啶、1,4-二氮杂二环[2.2.2]辛烷、1,8-二氮杂二环[5.4.0]十一碳-7-烯、四甲基胍、三乙烯二胺、四甲基乙二胺、n-甲基吗啉或n,n,n',n”,n
”‑
五甲基二亚乙基三胺。在本发明中,所述有机碱作为光氧化还原反应的电子给体,同时中和反应中生成的酸。
[0036]
在本发明中,所述第一有机溶剂优选为正己烷、环己烷、庚烷、苯、甲苯、二甲苯、三甲苯、三氟甲苯、氯苯、四氯化碳、氯仿、二氯甲烷、1,2-二氯乙烷、四氢呋喃、乙醚、汽油、二硫化碳、氯丙烷、溴乙烷、异丙醚、硝基甲烷、乙酸丁酯、正戊烷、二氧六环、乙酸甲酯、甲基叔丁基醚、石油醚、丙酮、乙腈、甲醇、乙醇、异丙醇、正丙醇、正丁醇、异丁醇、叔丁醇、水、吡啶、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲基亚砜、乙酸乙酯、三氟乙醇或六氟异丙醇。
[0037]
在本发明中,所述具有式i所示结构的联烯底物和光催化剂的摩尔比优选为1:(0.001~1),更优选为1:(0.01~0.9),进一步优选为1:(0.03~0.3);所述具有式i所示结构的联烯底物和有机碱的摩尔比优选为1:(1~20),更优选为1:(2~15),更优选为1:(3~10)。
[0038]
本发明对所述第一有机溶剂的用量没有特殊要求,能够将所述具有式i所示结构的联烯底物、光催化剂和有机碱完全溶解即可。
[0039]
在本发明中,所述自由基串联反应的温度优选为-50℃~100℃,更优选为-30℃~80℃,进一步优选为-20℃~20℃;光照波长优选为300nm~700nm,更优选为400nm~600nm;
反应时间优选为10~30h,更优选为13~25h,进一步优选为15~20h。
[0040]
在本发明中,所述自由基串联反应优选在空气氛围或保护气氛下进行。在本发明的实施例中,是在氮气气氛下进行。
[0041]
在本发明中,所述自由基串联反应的方程式如式1所示:
[0042][0043]
完成所述自由基串联反应后,本发明优选将所得反应产物体系进行减压浓缩,除去第一有机溶剂,得到具有式ii所示结构的自由基串联反应产物。本发明对所述减压浓缩的过程没有特殊要求,能够将第一有机溶剂除去即可。
[0044]
得到具有式ii所示结构的自由基串联反应产物后,本发明将所述具有式ii所示结构的自由基串联反应产物和酸溶于第二有机溶剂中,发生曼尼希环化反应,得到具有式iii所示结构的吲哚咔唑类化合物,
[0045][0046]
在本发明中,所述酸优选为质子酸或路易斯酸;所述质子酸优选为盐酸、硫酸、磷酸、硝酸、硼酸、氟硼酸、苯磺酸、氢氟酸、氢溴酸、氢碘酸、高氯酸、高碘酸、甲酸、乙酸、丙酸、丁二酸、己二酸、庚酸、甲磺酸、三氟甲磺酸、苯甲磺酸、对甲苯磺酸、樟脑磺酸或苯甲酸;所述路易斯酸优选为氯化锌、三氯化铝、三氟化硼、四氟硼酸锂、三氟甲磺酸银或三氟甲磺酸锌。本发明对所述酸的浓度没有特殊要求。
[0047]
在本发明中,所述第二有机溶剂优选为正己烷、环己烷、庚烷、苯、甲苯、二甲苯、三甲苯、三氟甲苯、氯苯、四氯化碳、氯仿、二氯甲烷、1,2-二氯乙烷、四氢呋喃、乙醚、汽油、二硫化碳、氯丙烷、溴乙烷、异丙醚、硝基甲烷、乙酸丁酯、正戊烷、二氧六环、乙酸甲酯、甲基叔丁基醚、石油醚、丙酮、乙腈、甲醇、乙醇、异丙醇、正丙醇、正丁醇、异丁醇、叔丁醇、水、吡啶、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲基亚砜、乙酸乙酯、三氟乙醇或六氟异丙醇。
[0048]
在本发明中,所述具有式i所示结构的联烯底物和酸的摩尔比优选为1:(1~200),更优选为1:(30~150),进一步优选为1:(50~100)。
[0049]
本发明对所述第二有机溶剂的用量没有特殊要求,能够将自由基串联反应产物完全溶解即可。
[0050]
在本发明中,所述曼尼希环化反应的温度优选为-20℃~130℃,更优选为0~100℃,进一步优选为20~70℃;时间优选为6~12h,更优选为7~10h。
[0051]
在本发明中,所述曼尼希环化反应的方程式如式2所示。
(dtbpy)]pf6(0.016毫摩尔)、n,n-二异丙基乙胺(1.6毫摩尔)溶于乙腈(8毫升)中,加毕后将反应物置于-15℃的低温条件下,在405nm光下照射反应16小时后减压浓缩;将其浓缩液溶于乙酸乙酯(6毫升),再加入37%浓盐酸(2毫升,24毫摩尔),加毕后将反应物置于50℃的油浴中继续反应10小时,用饱和碳酸钠水溶液(15毫升)淬灭反应,水相用乙酸乙酯萃取三次(15毫升
×
3),合并有机相,依次用水、饱和食盐水洗,无水硫酸钠干燥,浓缩后柱层析,得到吲哚咔唑化合物2(49.9毫克,76%产率)。
[0062]1h nmr(500mhz,acetone-d6)δ10.34(br.s,1h),7.32(d,j=8.0hz,1h),7.03(t,j=7.8hz,1h),7.00
–
6.95(m,2h),6.66(d,j=8.0hz,1h),6.55(d,j=7.8hz,1h),4.93(d,j=7.8hz,1h),3.59(ddd,j=11.8,7.8,4.2hz,1h),3.33(dt,j=15.9,4.2hz,1h),2.91(ddd,j=15.9,11.7,4.4hz,1h),2.32
–
2.24(m,1h),1.85(ddd,j=24.2,11.7,4.4hz,1h);
13
c nmr(150mhz,acetone-d6)δ152.6,137.9,134.6,130.0,129.8,129.1,125.7,123.9,121.9,119.2,118.1,110.7,110.0,107.8,54.6,41.5,24.5,21.5;hr-esi-ms(m/z):calcd.for c
18h15
cl2n2[m+h]
+
329.0607,found 329.0604.结果表明确实生成了化合物2。
[0063]
实施例3
[0064]
的合成。
[0065]
在氮气的氛围下,将相应底物(0.4毫摩尔)、光催化剂[ir(dfppy)2(dtbpy)]pf6(0.016毫摩尔)、n,n-二异丙基乙胺(1.6毫摩尔)溶于乙腈(8毫升)中,加毕后将反应物置于-15℃的低温条件下,在405nm光下照射反应16小时后减压浓缩;将其浓缩液溶于乙酸乙酯(6毫升),再加入37%浓盐酸(2毫升,24毫摩尔),加毕后将反应物置于50℃的油浴中继续反应10小时,用饱和碳酸钠水溶液(15毫升)淬灭反应,水相用乙酸乙酯萃取三次(15毫升
×
3),合并有机相,依次用水、饱和食盐水洗,无水硫酸钠干燥,浓缩后柱层析,得到吲哚咔唑化合物3(43.5毫克,68%产率)。
[0066]1hnmr(600mhz,acetone-d6)δ9.87(br.s,1h),6.93(m,3h),6.44(d,j=7.3hz,1h),6.32(d,j=8.2hz,1h),6.23(d,j=7.7hz,1h),4.82(d,j=7.9hz,1h),3.85(s,3h),3.83(s,3h),3.61
–
3.52(m,1h),3.05(dt,j=17.6,4.7hz,1h),2.83
–
2.78(m,1h),2.11
–
2.07(m,1h),1.98
–
1.91(m,1h);
13
c nmr(150mhz,acetone-d6)δ157.9,155.7,153.5,138.9,133.2,129.4,122.8,119.1,117.8,111.8,105.3,104.1,102.3,99.8,56.2,55.4,55.3,41.2,26.0,22.6;hr-esi-ms(m/z):calcd.for c
20h21
n2o2[m+h]
+
321.1598,found 321.1600.结果表明确实生成了化合物3。
[0067]
实施例4
[0068]
的合成。
[0069]
在氮气的氛围下,将相应底物(0.4毫摩尔)、光催化剂[ir(dfppy)2(dtbpy)]pf6(0.016毫摩尔)、n,n-二异丙基乙胺(1.6毫摩尔)溶于乙腈(8毫升)中,加毕后将反应物置于-15℃的低温条件下,在405nm光下照射反应16小时后减压浓缩;将其浓缩液溶于乙酸乙酯(6毫升),再加入37%浓盐酸(2毫升,24毫摩尔),加毕后将反应物置于50℃的油浴中继续反应10小时,用饱和碳酸钠水溶液(15毫升)淬灭反应,水相用乙酸乙酯萃取三次(15毫升
×
3),合并有机相,依次用水、饱和食盐水洗,无水硫酸钠干燥,浓缩后柱层析,得到吲哚咔唑化合物4(43.2毫克,73%产率)。1h nmr(600mhz,acetone-d6)δ10.02(br.s,1h),7.31(dd,j=8.6,4.4hz,1h),7.11(dd,j=8.8,2.4hz,1h),7.00(dd,j=8.8,2.4hz,1h),6.85(td,j=9.2,2.6hz,1h),6.67(td,j=9.2,2.6hz,1h),6.51(dd,j=8.6,4.4hz,1h),5.16(br.s,1h),4.95(d,j=8.0hz,1h),3.68(td,j=7.7,4.4hz,1h),2.66(t,j=6.0hz,2h),2.23
–
2.18(m,1h),2.11
–
2.06(m,1h);
19
f nmr(564mhz,acetone-d6)δ-127.0(s,1f),-128.4(s,1f);
13
c nmr(150mhz,acetone-d6)δ158.5(d,j
cf
=230.7hz);157.8(d,j
cf
=231.8hz);148.4,137.6,134.4,134.3,128.3(d,j
cf
=9.2hz),114.1(d,j
cf
=23.1hz),112.7(d,j
cf
=9.2hz),112.0(d,j
cf
=4.8hz),111.7(d,j
cf
=24.0hz),110.8(d,j
cf
=8.0hz),110.1(d,j
cf
=26.3hz),103.9(d,j
cf
=23.1hz),57.1,43.1,25.7,19.0;hr-esi-ms(m/z):calcd.for c
18h15
f2n2na[m+na]
+
297.1198,found 297.1197.结果表明确实生成了化合物4。
[0070]
实施例5
[0071]
的合成。
[0072]
在氮气的氛围下,将相应底物(0.4毫摩尔)、光催化剂[ir(dfppy)2(dtbpy)]pf6(0.016毫摩尔)、n,n-二异丙基乙胺(1.6毫摩尔)溶于乙腈(8毫升)中,加毕后将反应物置于-15℃的低温条件下,在405nm光下照射反应16小时后减压浓缩;将其浓缩液溶于乙酸乙酯(6毫升),再加入37%浓盐酸(2毫升,24毫摩尔),加毕后将反应物置于50℃的油浴中继续反应10小时,用饱和碳酸钠水溶液(15毫升)淬灭反应,水相用乙酸乙酯萃取三次(15毫升
×
3),合并有机相,依次用水、饱和食盐水洗,无水硫酸钠干燥,浓缩后柱层析,得到吲哚咔唑化合物5(54.5毫克,83%产率)。
[0073]1h nmr(600mhz,acetone-d6)δ10.09(br.s,1h),7.42(s,1h),7.33(d,j=8.6hz,1h),7.18(s,1h),7.04(d,j=8.6hz,1h),6.91(d,j=8.2hz,1h),6.52(d,j=8.2hz,1h),5.39(br.s,1h),4.98(d,j=8.0hz,1h),3.74
–
3.68(m,1h),2.68(t,j=6.0hz,2h),2.21(td,j=12.8,6.0hz,1h),2.09(dt,j=12.8,5.7hz,1h);
13
c nmr(150mhz,acetone-d6)δ150.1,136.1,135.2,133.5,128.1,127.1,124.0,123.5,122.1,121.3,117.7,112.2,110.8,110.4,55.8,41.8,24.8,18.1;hr-esi-ms(m/z):calcd.for c
18h15
cl2n2[m+h]
+
329.0607,found 329.0604.结果表明确实生成了化合物5。
[0074]
实施例6
[0075]
的合成。
[0076]
在氮气的氛围下,将相应底物(0.4毫摩尔)、光催化剂[ir(dfppy)2(dtbpy)]pf6(0.016毫摩尔)、n,n-二异丙基乙胺(1.6毫摩尔)溶于乙腈(8毫升)中,加毕后将反应物置于-15℃的低温条件下,在405nm光下照射反应16小时后减压浓缩;将其浓缩液溶于乙酸乙酯(6毫升),再加入37%浓盐酸(2毫升,24毫摩尔),加毕后将反应物置于50℃的油浴中继续反应10小时,用饱和碳酸钠水溶液(15毫升)淬灭反应,水相用乙酸乙酯萃取三次(15毫升
×
3),合并有机相,依次用水、饱和食盐水洗,无水硫酸钠干燥,浓缩后柱层析,得到吲哚咔唑化合物6(62.4毫克,75%产率)。
[0077]1h nmr(600mhz,acetone-d6)δ10.16(br.s,1h),7.58(s,1h),7.32(s,1h),7.31(d,j=8.6hz,1h),7.17(dd,j=8.6,1.7hz,1h),7.05(dd,j=8.2,1.3hz,1h),6.50(d,j=8.2hz,1h),5.47(br.s,1h),4.98(d,j=8.0hz,1h),3.76
–
3.68(m,1h),2.68(t,j=5.8hz,2h),2.23(td,j=12.9,5.8hz,1h),2.13
–
2.07(m,1h);
13
c nmr(150mhz,acetone-d6)δ151.5,136.8,136.3,135.0,130.9,129.8,127.3,124.9,121.7,113.7,112.5,112.0,111.6,110.0,56.7,42.7,25.8,19.0;hr-esi-ms(m/z):calcd.for c
18h15
br2n2[m+h]
+
416.9597,found 416.9593.结果表明确实生成了化合物6。
[0078]
实施例7
[0079]
的合成。
[0080]
在氮气的氛围下,将相应底物(0.4毫摩尔)、光催化剂[ir(dfppy)2(dtbpy)]pf6(0.016毫摩尔)、n,n-二异丙基乙胺(1.6毫摩尔)溶于乙腈(8毫升)中,加毕后将反应物置于-15℃的低温条件下,在405nm光下照射反应16小时后减压浓缩;将其浓缩液溶于乙酸乙酯(6毫升),再加入37%浓盐酸(2毫升,24毫摩尔),加毕后将反应物置于50℃的油浴中继续反应10小时,用饱和碳酸钠水溶液(15毫升)淬灭反应,水相用乙酸乙酯萃取三次(15毫升
×
3),合并有机相,依次用水、饱和食盐水洗,无水硫酸钠干燥,浓缩后柱层析,得到吲哚咔唑化合物7(44.4毫克,59%产率)。
[0081]1h nmr(600mhz,dmso-d6)δ11.11(s,1h),8.10(s,1h),7.72(s,2h),7.62(d,j=8.1hz,1h),7.44(d,j=8.5hz,1h),6.61
–
6.55(m,2h),4.99(d,j=8.2hz,1h),3.83(s,3h),3.77(s,3h),3.75
–
3.70(m,1h),2.70(dt,j=15.0,4.8hz,1h),2.61
–
2.53(m,1h),2.21(dd,
j=13.1,6.1hz,1h),2.14
–
2.00(m,1h);
13
c nmr(150mhz,dmso-d6)δ167.2,166.4,155.5,138.8,136.0,130.6,130.5,125.9,124.4,122.3,120.7,120.0,118.0,111.5,111.1,107.8,55.1,51.5,51.3,40.2,24.4,17.6;hr-esi-ms(m/z):calcd.for c
22h20
n2o4na[m+na]
+
399.1315,found 399.1317.结果表明确实生成了化合物7。
[0082]
实施例8
[0083]
的合成。
[0084]
在氮气的氛围下,将相应底物(0.4毫摩尔)、光催化剂[ir(dfppy)2(dtbpy)]pf6(0.016毫摩尔)、n,n-二异丙基乙胺(1.6毫摩尔)溶于乙腈(8毫升)中,加毕后将反应物置于-15℃的低温条件下,在405nm光下照射反应16小时后减压浓缩;将其浓缩液溶于乙酸乙酯(6毫升),再加入37%浓盐酸(2毫升,24毫摩尔),加毕后将反应物置于50℃的油浴中继续反应10小时,用饱和碳酸钠水溶液(15毫升)淬灭反应,水相用乙酸乙酯萃取三次(15毫升
×
3),合并有机相,依次用水、饱和食盐水洗,无水硫酸钠干燥,浓缩后柱层析,得到吲哚咔唑化合物8(40.3毫克,65%产率)。
[0085]1h nmr(600mhz,acetone-d6)δ10.58(br.s,1h),7.90(s,1h),7.52(m,2h),7.38(d,j=8.3hz,1h),7.30(d,j=8.0hz,1h),6.62(d,j=8.0hz,1h),6.27(br.s,1h),5.14(d,j=8.2hz,1h),3.87
–
3.79(m,1h),2.79
–
2.73(m,2h),2.34
–
2.27(m,1h),2.20
–
2.15(m,1h);
13
c nmr(150mhz,acetone-d6)δ155.9,139.4,137.3,134.1,132.8,128.0,127.8,125.4,124.8,121.3,121.0,113.2,113.1,109.9,102.9,100.5,56.3,42.1,25.6,18.8;hr-esi-ms(m/z):calcd.for c
20h15
n4[m+h]
+
311.1291,found 311.1294.结果表明确实生成了化合物8。
[0086]
实施例9
[0087]
的合成。
[0088]
在氮气的氛围下,将相应底物(0.4毫摩尔)、光催化剂[ir(dfppy)2(dtbpy)]pf6(0.016毫摩尔)、n,n-二异丙基乙胺(1.6毫摩尔)溶于乙腈(8毫升)中,加毕后将反应物置于-15℃的低温条件下,在405nm光下照射反应16小时后减压浓缩;将其浓缩液溶于乙酸乙酯(6毫升),再加入37%浓盐酸(2毫升,24毫摩尔),加毕后将反应物置于50℃的油浴中继续反应10小时,用饱和碳酸钠水溶液(15毫升)淬灭反应,水相用乙酸乙酯萃取三次(15毫升
×
3),合并有机相,依次用水、饱和食盐水洗,无水硫酸钠干燥,浓缩后柱层析,得到吲哚咔唑化合物9(35.7毫克,63%产率)。
[0089]1h nmr(600mhz,acetone-d6)δ9.74(br.s,1h),7.23
–
7.15(m,2h),7.00(s,1h),
6.88(d,j=8.4hz,1h),6.72(d,j=7.8hz,1h),6.43(d,j=7.8hz,1h),4.87(d,j=7.9hz,1h),3.64
–
3.55(m,1h),2.68
–
2.62(m,2h),2.36(s,3h),2.20(s,3h),2.19
–
2.14(m,1h),2.10
–
2.05(m,1h);
13
c nmr(150mhz,acetone-d6)δ149.8,136.0,135.6,132.6,128.5,128.2,128.2,127.8,124.9,123.6,118.8,111.5,111.2,110.4,56.7,43.0,26.2,21.6,21.0,19.4;hr-esi-ms(m/z):calcd.for c
20h21
n2[m+h]
+
289.1699,found 289.1694.结果表明确实生成了化合物9。
[0090]
实施例10
[0091]
的合成。
[0092]
在氮气的氛围下,将相应底物(0.4毫摩尔)、光催化剂[ir(dfppy)2(dtbpy)]pf6(0.016毫摩尔)、n,n-二异丙基乙胺(1.6毫摩尔)溶于乙腈(8毫升)中,加毕后将反应物置于-15℃的低温条件下,在405nm光下照射反应16小时后减压浓缩;将其浓缩液溶于乙酸乙酯(6毫升),再加入37%浓盐酸(2毫升,24毫摩尔),加毕后将反应物置于50℃的油浴中继续反应10小时,用饱和碳酸钠水溶液(15毫升)淬灭反应,水相用乙酸乙酯萃取三次(15毫升
×
3),合并有机相,依次用水、饱和食盐水洗,无水硫酸钠干燥,浓缩后柱层析,得到吲哚咔唑化合物10(42.3毫克,66%产率)。
[0093]1h nmr(600mhz,acetone-d6)δ9.70(br.s,1h),7.20(d,j=8.7hz,1h),6.92(d,j=1.3hz,1h),6.85(d,j=1.9hz,1h),6.70(dd,j=8.7,1.3hz,1h),6.52(dd,j=8.3,1.9hz,1h),6.46(d,j=8.3hz,1h),4.87(d,j=7.8hz,1h),4.83(br.s,1h),3.77(s,3h),3.70(s,3h),3.63
–
3.58(m,1h),2.69
–
2.62(m,2h),2.22
–
2.15(m,1h),2.10
–
2.06(m,1h);
13
c nmr(150mhz,acetone-d6)δ154.9,154.6,146.0,136.6,134.1,132.9,128.4,113.2,112.4,112.1,111.6,111.4,111.2,101.4,57.1,56.2,56.0,43.5,26.1,19.5;hr-esi-ms(m/z):calcd.for c
20h21
n2o2[m+h]
+
321.1598,found 321.1595.结果表明确实生成了化合物10。
[0094]
实施例11
[0095]
的合成。
[0096]
在氮气的氛围下,将相应底物(0.4毫摩尔)、光催化剂[ir(dfppy)2(dtbpy)]pf6(0.016毫摩尔)、n,n-二异丙基乙胺(1.6毫摩尔)溶于乙腈(8毫升)中,加毕后将反应物置于-15℃的低温条件下,在405nm光下照射反应16小时后减压浓缩;将其浓缩液溶于乙酸乙酯(6毫升),再加入37%浓盐酸(2毫升,24毫摩尔),加毕后将反应物置于50℃的油浴中继续反应10小时,用饱和碳酸钠水溶液(15毫升)淬灭反应,水相用乙酸乙酯萃取三
次(15毫升
×
3),合并有机相,依次用水、饱和食盐水洗,无水硫酸钠干燥,浓缩后柱层析,得到吲哚咔唑化合物11(37.9毫克,64%产率)。
[0097]1h nmr(500mhz,acetone-d6)δ10.04(br.s,1h),7.41
–
7.36(m,1h),7.16
–
7.11(m,1h),7.08(d,j=10.1hz,1h),6.79(t,j=9.2hz,1h),6.34(t,j=9.0hz,1h),6.29(d,j=10.2hz,1h),4.97(d,j=8.0hz,1h),3.66
–
3.58(m,1h),2.66(t,j=6.0hz,2h),2.21
–
2.14(m,1h),2.10
–
2.06(m,1h);
19
f nmr(564mhz,acetone-d6)δ-117.7(s,1f),-123.4(s,1f);
13
c nmr(150mhz,acetone-d6)δ164.3(d,j
cf
=232.7hz),160.7(d,j
cf
=232.9hz),153.9(d,j
cf
=12.2hz),137.7(d,j
cf
=12.3hz),135.8,127.9,124.9(d,j
cf
=10.2hz),124.8,120.0(d,j
cf
=10.1hz),112.0,107.8(d,j
cf
=24.3hz),104.4(d,j
cf
=22.8hz),98.1(d,j
cf
=25.5hz),97.8(d,j
cf
=26.3hz),57.1,41.9,26.2,19.2;hr-esi-ms(m/z):calcd.for c
18h15
f2n2[m+h]
+
297.1198,found 297.1196.结果表明确实生成了化合物11。
[0098]
实施例12
[0099]
的合成。
[0100]
在氮气的氛围下,将相应底物(0.4毫摩尔)、光催化剂[ir(dfppy)2(dtbpy)]pf6(0.016毫摩尔)、n,n-二异丙基乙胺(1.6毫摩尔)溶于乙腈(8毫升)中,加毕后将反应物置于-15℃的低温条件下,在405nm光下照射反应16小时后减压浓缩;将其浓缩液溶于乙酸乙酯(6毫升),再加入37%浓盐酸(2毫升,24毫摩尔),加毕后将反应物置于50℃的油浴中继续反应10小时,用饱和碳酸钠水溶液(15毫升)淬灭反应,水相用乙酸乙酯萃取三次(15毫升
×
3),合并有机相,依次用水、饱和食盐水洗,无水硫酸钠干燥,浓缩后柱层析,得到吲哚咔唑化合物12(49.2毫克,75%产率)。
[0101]1h nmr(500mhz,acetone-d6)δ10.12(br.s,1h),7.41(d,j=8.4hz,1h),7.37(d,j=1.8hz,1h),7.15(d,j=7.8hz,1h),6.98(dd,j=8.4,1.8hz,1h),6.62(dd,j=7.8,1.8hz,1h),6.54(d,j=1.8hz,1h),5.55(br.s,1h),4.98(d,j=7.9hz,1h),3.70
–
3.61(m,1h),2.67(t,j=6.0hz,2h),2.23
–
2.15(m,1h),2.11
–
2.06(m,1h);
13
c nmr(150mhz,acetone-d6)δ153.7,138.1,136.2,133.5,131.1,127.7,126.7,125.3,120.3,120.1,118.4,112.1,111.8,110.2,56.8,42.1,25.9,19.1;hr-esi-ms(m/z):calcd.for c
18h15
cl2n2[m+h]
+
329.0607,found 329.0605.结果表明确实生成了化合物12。
[0102]
实施例13
[0103]
的合成。
[0104]
在氮气的氛围下,将相应底物(0.4毫摩尔)、光催化剂[ir(dfppy)2(dtbpy)]pf6(0.016毫摩尔)、n,n-二异丙基乙胺(1.6毫摩尔)溶于乙腈(8毫升)中,加毕后将反应物置于-15℃的低温条件下,在405nm光下照射反应16小时后减压浓缩;将其浓缩液溶于乙酸乙酯(6毫升),再加入37%浓盐酸(2毫升,24毫摩尔),加毕后将反应物置于50℃的油浴中继续反应10小时,用饱和碳酸钠水溶液(15毫升)淬灭反应,水相用乙酸乙酯萃取三次(15毫升
×
3),合并有机相,依次用水、饱和食盐水洗,无水硫酸钠干燥,浓缩后柱层析,得到吲哚咔唑化合物13(56.6毫克,68%产率)。
[0105]1h nmr(600mhz,acetone-d6)δ10.03(br.s,1h),7.47(s,1h),7.30(d,j=8.4hz,1h),7.06(d,j=8.4hz,1h),7.03(d,j=7.8hz,1h),6.72(d,j=7.8hz,1h),6.64(s,1h),5.47(br.s,1h),4.91(d,j=8.0hz,1h),3.61
–
3.54(m,1h),2.60(t,j=5.9hz,2h),2.15
–
2.08(m,1h),2.04
–
2.00(m,1h);
13
c nmr(150mhz,acetone-d6)δ153.9,138.6,136.1,131.6,127.0,125.8,122.7,121.5,121.4,120.8,115.4,114.8,113.1,112.2,56.7,42.2,25.9,19.1;hr-esi-ms(m/z):calcd.for c
18h15
br2n2[m+h]
+
416.9597,found 416.9597.结果表明确实生成了化合物13。
[0106]
实施例14
[0107]
的合成。
[0108]
在氮气的氛围下,将相应底物(0.4毫摩尔)、光催化剂[ir(dfppy)2(dtbpy)]pf6(0.016毫摩尔)、n,n-二异丙基乙胺(1.6毫摩尔)溶于乙腈(8毫升)中,加毕后将反应物置于-15℃的低温条件下,在405nm光下照射反应16小时后减压浓缩;将其浓缩液溶于乙酸乙酯(6毫升),再加入37%浓盐酸(2毫升,24毫摩尔),加毕后将反应物置于50℃的油浴中继续反应10小时,用饱和碳酸钠水溶液(15毫升)淬灭反应,水相用乙酸乙酯萃取三次(15毫升
×
3),合并有机相,依次用水、饱和食盐水洗,无水硫酸钠干燥,浓缩后柱层析,得到吲哚咔唑化合物14(35.2毫克,61%产率)。
[0109]1h nmr(500mhz,acetone-d6)δ9.73(br.s,1h),7.27(d,j=8.0hz,1h),7.11(s,1h),7.03(d,j=7.4hz,1h),6.80(d,j=8.0hz,1h),6.45(d,j=7.4hz,1h),6.35(s,1h),4.87(d,j=7.9hz,1h),3.62
–
3.54(m,1h),2.64(t,j=6.0hz,2h),2.37(s,3h),2.21
–
2.15(m,1h),2.14(s,3h),2.04
–
1.99(m,1h);
13
c nmr(150mhz,acetone-d6)δ152.5,138.2,137.7,135.0,131.5,129.5,126.0,123.9,121.2,119.6,118.8,111.8,111.6,111.4,56.8,42.6,26.2,21.9,21.6,19.4;hr-esi-ms(m/z):calcd.for c
20h21
n2[m+h]
+
289.1699,found 289.1701.结果表明确实生成了化合物14。
[0110]
实施例15
[0111]
的合成。
[0112]
在氮气的氛围下,将相应底物(0.4毫摩尔)、光催化剂[ir(dfppy)2(dtbpy)]pf6(0.016毫摩尔)、n,n-二异丙基乙胺(1.6毫摩尔)溶于乙腈(8毫升)中,加毕后将反应物置于-15℃的低温条件下,在405nm光下照射反应16小时后减压浓缩;将其浓缩液溶于乙酸乙酯(6毫升),再加入37%浓盐酸(2毫升,24毫摩尔),加毕后将反应物置于50℃的油浴中继续反应10小时,用饱和碳酸钠水溶液(15毫升)淬灭反应,水相用乙酸乙酯萃取三次(15毫升
×
3),合并有机相,依次用水、饱和食盐水洗,无水硫酸钠干燥,浓缩后柱层析,得到吲哚咔唑化合物15(41.6毫克,65%产率)。
[0113]1h nmr(600mhz,acetone-d6)δ9.69(br.s,1h),7.26(d,j=8.5hz,1h),7.02(d,j=8.0hz,1h),6.86(d,j=2.0hz,1h),6.63(dd,j=8.5,1.9hz,1h),6.19(dd,j=8.0,2.0hz,1h),6.13(d,j=1.9hz,1h),5.14(br.s,1h),4.87(d,j=7.9hz,1h),3.75(s,3h),3.64(s,3h),3.57
–
3.52(m,1h),2.64
–
2.59(m,2h),2.16
–
2.10(m,1h),2.03
–
1.98(m,1h);
13
c nmr(150mhz,acetone-d6)δ161.1,157.2,153.4,138.4,134.1,124.5,124.2,122.3,119.4,111.5,109.2,103.7,97.2,95.5,56.9,55.7,55.3,41.9,26.2,19.2;hr-esi-ms(m/z):calcd.for c
20h21
n2o2[m+h]
+
321.1598,found 321.1597.结果表明确实生成了化合物15。
[0114]
实施例16
[0115]
的合成。
[0116]
在氮气的氛围下,将相应底物(0.4毫摩尔)、光催化剂[ir(dfppy)2(dtbpy)]pf6(0.016毫摩尔)、n,n-二异丙基乙胺(1.6毫摩尔)溶于乙腈(8毫升)中,加毕后将反应物置于-15℃的低温条件下,在405nm光下照射反应16小时后减压浓缩;将其浓缩液溶于乙酸乙酯(6毫升),再加入37%浓盐酸(2毫升,24毫摩尔),加毕后将反应物置于50℃的油浴中继续反应10小时,用饱和碳酸钠水溶液(15毫升)淬灭反应,水相用乙酸乙酯萃取三次(15毫升
×
3),合并有机相,依次用水、饱和食盐水洗,无水硫酸钠干燥,浓缩后柱层析,得到吲哚咔唑化合物16(52.1毫克,88%产率)。
[0117]1h nmr(500mhz,acetone-d6)δ10.27(br.s,1h),7.25(d,j=7.8hz,1h),7.06(d,j=7.3hz,1h),6.98
–
6.92(m,1h),6.88
–
6.82(m,1h),6.82
–
6.72(m,1h),6.70
–
6.64(m,1h),5.36(br.s,1h),5.08(d,j=8.0hz,1h),3.80
–
3.73(m,1h),2.71(t,j=5.8hz,2h),2.27
–
2.19(m,1h),2.19
–
2.08(m,1h);
19
f nmr(564mhz,acetone-d6)δ-137.0(s,1f),-138.0(s,1f);
13
c nmr(150mhz,acetone-d6)δ151.2(d,j
cf
=57.6hz),149.6(d,j
cf
=54.5hz),138.6
(d,j
cf
=12.8hz),136.3(d,j
cf
=4.7hz),136.2,131.8(d,j
cf
=5.4hz),125.4(d,j
cf
=12.9hz),120.3(d,j
cf
=2.7hz),120.1(d,j
cf
=6.0hz),119.9(d,j
cf
=5.7hz),115.4(d,j
cf
=3.0hz),114.9(d,j
cf
=17.6hz),113.0,107.2(d,j
cf
=16.4hz),57.2,43.2,25.8,19.4;hr-esi-ms(m/z):calcd.for c
18h15
f2n2[m+h]
+
297.1198,found 297.1194.结果表明确实生成了化合物16。
[0118]
实施例17
[0119]
的合成。
[0120]
在氮气的氛围下,将相应底物(0.4毫摩尔)、光催化剂[ir(dfppy)2(dtbpy)]pf6(0.016毫摩尔)、n,n-二异丙基乙胺(1.6毫摩尔)溶于乙腈(8毫升)中,加毕后将反应物置于-15℃的低温条件下,在405nm光下照射反应16小时后减压浓缩;将其浓缩液溶于乙酸乙酯(6毫升),再加入37%浓盐酸(2毫升,24毫摩尔),加毕后将反应物置于50℃的油浴中继续反应10小时,用饱和碳酸钠水溶液(15毫升)淬灭反应,水相用乙酸乙酯萃取三次(15毫升
×
3),合并有机相,依次用水、饱和食盐水洗,无水硫酸钠干燥,浓缩后柱层析,得到吲哚咔唑化合物17(51.8毫克,79%产率)。
[0121]1h nmr(600mhz,acetone-d6)δ10.24(br.s,1h),7.41(d,j=7.8hz,1h),7.18
–
7.10(m,2h),7.04
–
6.96(m,2h),6.67(t,j=7.9hz,1h),5.57(br.s,1h),5.07(d,j=7.9hz,1h),3.85
–
3.69(m,1h),2.71(t,j=5.9hz,2h),2.23
–
2.16(m,1h),2.15
–
2.08(m,1h);
13
c nmr(125mhz,acetone-d6)δ148.8,136.1,134.6,134.2,129.6,128.1,123.1,121.9,120.8,120.2,118.3,116.9,115.3,113.3,56.3,43.6,26.0,19.5;hr-esi-ms(m/z):calcd.for c
18h15
cl2n2[m+h]
+
329.0607,found 329.0603.结果表明确实生成了化合物17。
[0122]
实施例18
[0123]
的合成。
[0124]
在氮气的氛围下,将相应底物(0.4毫摩尔)、光催化剂[ir(dfppy)2(dtbpy)]pf6(0.016毫摩尔)、n,n-二异丙基乙胺(1.6毫摩尔)溶于乙腈(8毫升)中,加毕后将反应物置于-15℃的低温条件下,在405nm光下照射反应16小时后减压浓缩;将其浓缩液溶于乙酸乙酯(6毫升),再加入37%浓盐酸(2毫升,24毫摩尔),加毕后将反应物置于50℃的油浴中继续反应10小时,用饱和碳酸钠水溶液(15毫升)淬灭反应,水相用乙酸乙酯萃取三次(15毫升
×
3),合并有机相,依次用水、饱和食盐水洗,无水硫酸钠干燥,浓缩后柱层析,得
到吲哚咔唑化合物18(46.2毫克,72%产率)。
[0125]1h nmr(600mhz,acetone-d6)δ9.98(br.s,1h),7.02(d,j=7.7hz,1h),6.90(t,j=7.7hz,1h),6.87
–
6.82(m,1h),6.66
–
6.60(m,3h),5.10(br.s,1h),4.97(d,j=7.8hz,1h),3.91(s,3h),3.71(s,3h),3.70
–
3.63(m,1h),2.67(t,j=5.6hz,2h),2.24
–
2.16(m,1h),2.12
–
2.06(m,1h);
13
c nmr(150mhz,acetone)δ147.2,146.3,140.9,135.2,132.9,129.3,127.7,120.1,119.6,117.0,112.3,112.1,110.6,102.8,57.1,55.6,55.6,43.5,26.0,19.7;hr-esi-ms(m/z):calcd.for c
20h21
n2o2[m+h]
+
321.1598,found 321.1597.结果表明确实生成了化合物18。
[0126]
实施例19
[0127]
的合成。
[0128]
在氮气的氛围下,将相应底物(0.4毫摩尔)、光催化剂[ir(dfppy)2(dtbpy)]pf6(0.016毫摩尔)、n,n-二异丙基乙胺(1.6毫摩尔)溶于乙腈(8毫升)中,加毕后将反应物置于-15℃的低温条件下,在405nm光下照射反应16小时后减压浓缩;将其浓缩液溶于乙酸乙酯(6毫升),再加入37%浓盐酸(2毫升,24毫摩尔),加毕后将反应物置于50℃的油浴中继续反应10小时,用饱和碳酸钠水溶液(15毫升)淬灭反应,水相用乙酸乙酯萃取三次(15毫升
×
3),合并有机相,依次用水、饱和食盐水洗,无水硫酸钠干燥,浓缩后柱层析,得到吲哚咔唑化合物19(45.4毫克,63%产率)。
[0129]1h nmr(600mhz,dmso-d6)δ10.69(br.s,1h),7.91
–
7.85(m,3h),7.81(s,1h),7.70
–
7.65(m,2h),7.49(d,j=8.1hz,1h),7.28(t,j=7.4hz,1h),7.23(t,j=7.4hz,2h),7.12(t,j=7.4hz,1h),6.79(s,1h),5.02(d,j=7.6hz,1h),3.86(dd,j=14.9,5.8hz,1h),2.80(dt,j=14.9,4.9hz,1h),2.72
–
2.61(m,1h),2.46
–
2.37(m,1h),2.23
–
2.13(m,1h);
13
c nmr(125mhz,dmso-d6)δ149.5,139.4,137.0,134.5,134.2,129.7,128.9,128.1,127.8,127.7,127.4,127.2,125.4,125.2,123.1,121.9,121.7,121.5,115.1,109.0,105.9,101.9,55.2,40.5,24.2,17.7;hr-esi-ms(m/z):calcd.for c
26h21
n2[m+h]
+
361.1699,found 361.1701.结果表明确实生成了化合物19。
[0130]
由以上实施例可知,本发明提供了一种吲哚咔唑类化合物的制备方法,以联烯底物为原料,通过光激发发生单电子转移引发自由基串联反应,并在酸性条件下发生曼尼希环化一锅合成吲哚咔唑类化合物,方法简单、高效,原料和试剂便宜易得,反应产率高,副产物少,反应的化学和区域选择性高;本发明方法操作简单,适于工业化生产和市场推广应用。
[0131]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1