一种1,2,4-三唑环的1,4萘醌类STAT3抑制剂及其应用
一种1,2,4-三唑环的1,4萘醌类stat3抑制剂及其应用
技术领域
1.本发明涉及stat3抑制剂技术领域,具体涉及一种1,2,4-三唑环的1,4萘醌类stat3抑制剂及其应用。
背景技术:
2.signal transducer and activator of transcription 3(stat3)是典型的stat家族的成员,stat家族是响应于细胞因子/生长因子而被激活以促进增殖、存活和其它生物学过程的潜在转录因子。stat3的结构主要由一个n-端域、卷曲螺旋域、dna结合域、sh2结构域和一个c-端反式激活域。其中,dna结合域和sh2结构域的作用最重要。sh2区是stat3蛋白中最为保守的一个结构域,对stat3磷酸化二聚体的形成至关重要。tyr705和ser727是磷酸化位点,lys685为乙酰化位点。
3.在许多癌症中,stat3在肿瘤的生长和转移中起着重要的作用,包括细胞增殖、入侵、迁移和血管生成。受体酪氨酸激酶和/或src家族激酶等介导的关键酪氨酸残基可以磷酸化激活stat3,但主要被白介素(il-6)家族细胞因子受体相关的janus激酶(jaks)激活,通过酪氨酸磷酸化,stat3形成同源二聚体并易位到核,与靶基因的启动子中的特异性dna-效应元件结合,并诱导基因表达。
4.近年来,有众多的stat3抑制剂被研究,但最终都因为药物稳定性差、生物利用度差、药代动力学性质差和膜渗透性差等原因未能进入临床或晚期临床研究。sta-21是第一个小分子stat3抑制剂,有效的抑制了乳腺癌肿瘤细胞系的生长和存活,比如表达持续活化stat的3mda-mb-231,mda-mb-435s以及mda-mb-468细胞。在rh30和rd2细胞中,sta-21通过caspases 3、8、9的调节抑制了细胞的活性和生长,同时诱导了细胞的凋亡。sta-21在2011年完成了1/2期临床研究,后续未见深入的临床研究结果。专利cn101854937提供了一种1,4-萘醌结构用于stat3抑制剂。该专利提供的核心化合物(代号:bbi608)也具有一些临床缺陷。结构刚性大,油水溶解性差,导致口服吸收差、生物利用度低。
5.因此,目前市面上还没有一种安全有效类药性好的stat3抑制剂。
技术实现要素:
6.针对现有技术中的缺陷,本发明提供一种1,2,4-三唑环的1,4萘醌类stat3抑制剂及其应用。本发明通过的1,2,4-三唑环的1,4萘醌类stat3抑制剂,可有效降低egf激活的stat3磷酸化修饰,同时可有效抑制il-6刺激介导的stat3磷酸化修饰,安全有效活性高,可有效降低nanog的转录水平。
7.本发明的一个目的在于保护一种1,2,4-三唑环的1,4萘醌类stat3抑制剂。所述的1,2,4-三唑环的1,4萘醌类stat3抑制剂包括如式(1)、式(2)、式(3)、式(4)的化合物,式(1)、式(2)、式(3)、式(4)的化合物的互变异构体,立体异构体,混合物,水合物,溶剂化物,晶型盐,代谢产物,可药用的盐中的一种或多种;
8.式(1)为式(2)为
9.式(3)为式(4)为
10.其中,式(1)、式(2)、式(3)、式(4)中r均为h、c1-c4的烷基、苯基、苄基、取代苯基、取代苄基、三氟甲基、2-甲氧基苯基、4-甲氧基苯基、2,3,5-三甲氧基苯基、4-甲基苯基、吡啶基中的一种;r1均为h、c1-c4的烷基中的一种。
11.优选地,式(1)中,r1为h,r为h、甲基、乙基、丙基、异丙基、苯基、苄基、三氟甲基、2-甲氧基苯基、4-甲氧基苯基、2,3,5-三甲氧基苯基、4-甲基苯基、吡啶基中的一种。
12.优选地,式(2)中,r1为h,r为h、甲基、乙基、丙基、异丙基、苯基、苄基、三氟甲基、2-甲氧基苯基、4-甲氧基苯基、2,3,5-三甲氧基苯基、4-甲基苯基、吡啶基中的一种。
13.优选地,式(3)中,r1为h,r为h、甲基、乙基、丙基、异丙基、苯基、苄基、三氟甲基、2-甲氧基苯基、4-甲氧基苯基、2,3,5-三甲氧基苯基、4-甲基苯基、吡啶基中的一种。
14.优选地,式(4)中,r1为h,r为h、甲基、乙基、丙基、异丙基、苯基、苄基、三氟甲基、2-甲氧基苯基、4-甲氧基苯基、2,3,5-三甲氧基苯基、4-甲基苯基、吡啶基中的一种。
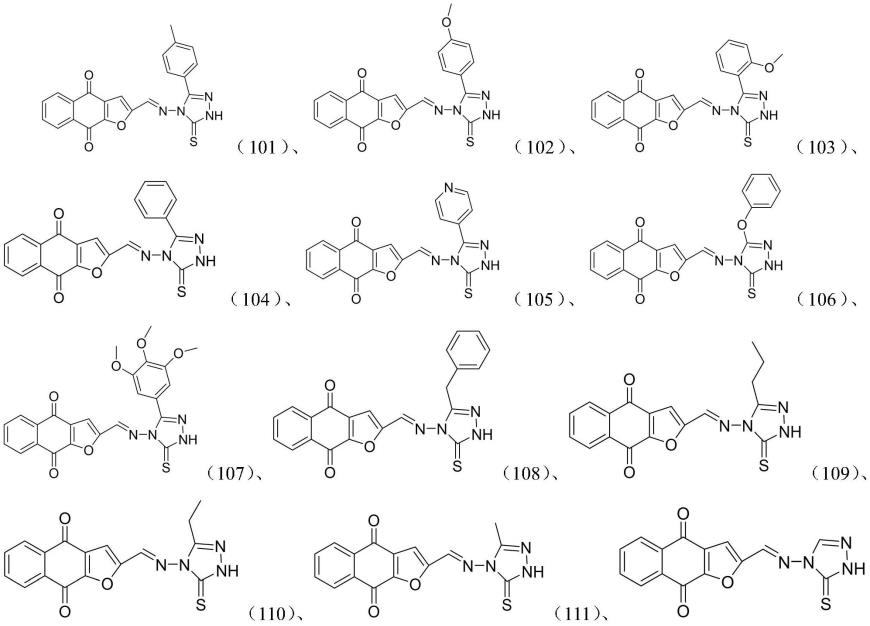
15.优选地,所述的1,2,4-三唑环的1,4萘醌类stat3抑制剂包括:
16.17.[0018][0019]
中的一种或多种。
[0020]
本发明的另一个目的在于保护上述的1,2,4-三唑环的1,4萘醌类stat3抑制剂用于制备药物的应用,所述的药物为治疗癌症或与异常的stat3途径活性有关的紊乱的疾病的药物。
[0021]
本发明的有益效果体现在:
[0022]
(1)本发明提供的1,2,4-三唑环的1,4萘醌类stat3抑制剂可有效降低egf激活的stat3磷酸化修饰,同时可有效抑制il-6刺激介导的stat3磷酸化修饰,并且这一作用具有剂量依赖性。
[0023]
(2)本发明提供的1,2,4-三唑环的1,4萘醌类stat3抑制剂安全有效,可有效降低nanog的转录水平,相较于专利cn101854937中提供的化合物bbi608具有更好的活性和类药性。
附图说明
[0024]
为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍。在所有附图中,类似的元件或部分一般由类似的附图标记标识。附图中,各元件或部分并不一定按照实际的比例绘制。
[0025]
图1为本发明实施例1中化合物d的合成路线图;
[0026]
图2为本发明实施例1中化合物h的合成路线图;
[0027]
图3为本发明实施例1中化合物101的合成路线图;
[0028]
图4为本发明实施例1中化合物102的合成路线图;
[0029]
图5为本发明实施例1中化合物108的合成路线图;
[0030]
图6为dz-1抑制hela细胞中egf介导的stat3磷酸化水平检测图;
[0031]
图7为dz-1抑制在肝癌细胞hepg2中il-6介导的stat3磷酸化水平检测图;
[0032]
图8为dz-1抑制人神经胶质瘤细胞u87中il-6介导的stat3磷酸化水平检测图;
[0033]
图9为dz-1抑制人神经母瘤细胞ln229中il-6介导的stat3磷酸化水平检测图;
[0034]
图10为dz-1抑制stat3下游nanog基因的转录的检测图;
[0035]
图11为dz-1抑制stat3下游oct4基因的转录的检测图;
[0036]
图12dz-1抑制stat3下游β-catenin基因的转录检测图。
具体实施方式
[0037]
下面将结合附图对本发明技术方案的实施例进行详细的描述。以下实施例仅用于更加清楚地说明本发明的技术方案,因此只作为示例,而不能以此来限制本发明的保护范围。
[0038]
需要注意的是,除非另有说明,本技术使用的技术术语或者科学术语应当为本发明所属领域技术人员所理解的通常意义。
[0039]
实施例1
[0040]
本实施例提供了化合物101的合成方法,如图1、图2和图3所示,步骤如下:
[0041]
步骤s1.4,9-dioxo-4,9-dihydronaphtho[2,3-b]furan-2-carbaldehyde的合成:将2-羟基-1,4-萘醌(2.06g,11.8mmol),记为化合物a,溶解于40ml二氯甲烷,碘苯二乙酯(3.8g)溶于二氯甲烷(25ml)在100ml的反应瓶中,反应瓶置于0℃条件下加入含有2-羟基-1,4-萘醌的二氯甲烷溶液,混合液在0℃温度下反应1h,然后在室温继续反应4h。过滤后得到橘色沉淀,沉淀用二氯甲烷洗涤、干燥得到化合物b。将化合物b(4.0g,10.8mmol)溶于160ml吡啶中,炔丙醇(6.4ml,108mmol)和氧化亚铜(4.0g)加入到上述溶液中,在80℃的条件下反应2h。溶液过滤并用10%的稀盐酸稀释。酸性溶液用乙酸乙酯萃取三次,合并的有机相用水洗,无水硫酸钠干燥。tlc薄层(氯仿/丙酮)过柱子得到橘色固体化合物c。将化合物c(600mg,2.6mmol)溶于30ml二氯甲烷中,加入pcc(2.0g)。混合液在室温条件下反应20h,用水稀释然后用乙酸乙酯萃取3次,合并的有机相用水洗涤,无水硫酸钠干燥。真空除去溶剂,薄层色谱(氯仿/丙酮)分离得到化合物d。
[0042]
将化合物d进行核磁共振氢谱检测,结果如下:1h nmr(400hz,dmso-d6):9.91(s,1h),8.15(s,2h),8.04(s,1h),7.93(s,2h)。
[0043]
步骤s2.4-amino-5-(p-tolyl)-2,4-dihydro-3h-1,2,4-triazole-3-thione的合成:将对甲基苯甲酸乙酯(16.4g,0.1mol),记为化合物e,溶于60ml无水乙醇,分液漏斗滴入水合肼30ml,磁力搅拌器设置100℃,搅拌反应,tlc检测直到原料反应完全,减压浓缩,残渣溶于60ml水,用乙酸乙酯萃取60ml
×
4次,合并萃取液,无水硫酸镁干燥,过滤,除去硫酸镁固体,液体浓缩得纯品化合物f。在三颈烧瓶中加入化合物f(3.0g,2.0mmol),koh(0.168g,),无水乙醇14ml搅拌使其完全溶解,缓慢滴加0.18ml cs2至h2s气体完全放出,减压浓缩得化合物g。将1mmol化合物g和85%水合肼混合均匀,80ml无水乙醇倒入反应瓶中,
磁力搅拌器设置100℃,搅拌6h。冷却至室温,倒入冰水中,稀盐酸调至弱酸性,析出晶体,过滤,滤饼干燥后用无水乙醇重结晶得白色晶体化合物h。
[0044]
步骤s3.化合物101的合成:将2mmol化合物d与2mmol化合物h加入到50ml的烧杯中,随后加入30ml乙醇和2ml冰乙酸,100℃回流8h,真空除去溶剂,残渣用薄层色谱(石油醚/乙酸乙酯)分离得到化合物101,产率74%。
[0045]
将实施例1制得的化合物101进行核磁共振氢谱检测,结果如下:1h nmr(400mhz,[d6]dmso):δ14.27(s,1h),10.12(s,1h),8.10(m,2h),7.89(m,3h),7.73(d,j=8hz,2h),7.32(d,j=8hz,2h),2.35(s,3h).
13
c nmr(400mhz,[d6]dmso):δ180.14,173.85,162.72,154.31,152.57,152.52,149.63,141.10,135.00,133.34,133.05,131.04,129.83,129.01,127.20,127.03,122.93,115.75,21.55.ms:437.32(m+na)。
[0046]
实施例2
[0047]
本实施例提供了化合物102的合成方法,如图4所示,步骤如下:将3mmol实施例1制得的化合物d与3mmol 4-氨基-5-(4-甲氧基苯基)-1,2,4-三唑-3-硫酮投入到100ml的反应瓶,随后加入40ml冰乙酸,120℃回流4h,真空减压除去溶剂,残渣用薄层色谱(石油醚/乙酸乙酯)分离得到化合物102,产率75%。
[0048]
将实施例2制得的化合物102进行核磁共振氢谱检测,结果如下:1h nmr(400mhz,[d6]dmso):δ14.21(s,1h),10.10(s,1h),8.11(m,2h),7.90(m,3h),7.81(s,1h),7.79(s,1h),7.07(m,2h),3.80(s,3h).
13
c nmr(400mhz,[d6]dmso):δ180.14,173.87,162.58,161.72,160.99,154.32,152.73,152.55,149.44,135.00,133.36,133.06,131.07,130.69,130.11,127.21,127.04,117.97,115.68,114.77,114.48,55.96.ms:453.22(m+na)。
[0049]
实施例3
[0050]
本实施例提供了化合物108的合成方法,如图5所示,步骤如下:将1mmol实施例1制得的化合物d与1mmol 4-氨基-5-苯基-1,2,4-三唑-3-硫酮投入到50ml的反应瓶,随后加入20ml乙醇和1ml冰乙酸,100℃回流6h,真空除去溶剂,残渣用薄层色谱(石油醚/乙酸乙酯)分离得到化合物8,产率83%。
[0051]
将实施例3制得的化合物108进行核磁共振氢谱检测,结果如下:1h nmr(400mhz,[d6]dmso):δ14.01(s,1h),10.57(s,1h),8.15(m,2h),7.89(m,3h),7.73(m,5h),4.18(s,2h).ms:437.32(m+na)。
[0052]
试验例1
[0053]
gtc细胞活性筛选
[0054]
受试细胞株如表1所示。配制完全培养基,充分混匀,复苏细胞,传至少两代后左右选择生长状态良好的细胞株。将细胞培养瓶从培养箱中取出,吸掉培养基,用胰酶洗一遍,弃掉废液,加3ml新鲜胰酶于培养瓶消化。待细胞松动要脱离瓶壁时,加8ml完全培养基中止胰酶消化,并轻轻混匀。用移液管将细胞悬液移入离心管中,800-1000rpm的转速离心3-5min。悬浮细胞:吸取细胞悬液并移入离心管中,800-1000rpm的转速离心3-5min,弃上清,向离心管中加适当体积的培养基,轻柔吹打使细胞重悬均匀,使用vi-cell xr细胞计数仪计数,将细胞悬液调至合适浓度,将细胞悬液加入96孔板中,100μl/孔。标记细胞名称,种板密度,日期等详细信息,将培养板放置于co2培养箱中过夜。
[0055]
表1首次受试细胞株
[0056]
细胞株接种密度(细胞数/孔)完全培养基nci-h12993500rpmi1640+10%fbsmcf73500dmem+10%fbsmiapaca-23500rpmi1640+10%fbs
[0057]
室温融化celltiter-glo buffer。将冻干celltiter glo底物平衡至室温,将celltiter-glo buffer加入celltiter glo底物中并充分混匀。将加入药物的细胞板取出平衡至室温。每孔中加入混匀后的celltiter glo试剂100μl,避光振荡2min,孵育10min。将培养板放入enspire读板,记录luminescence读值结果;按下列公式计算抑制率:抑制率(%)=(1-(rlu compound-rlu blank))/(rlu dmso-rlu blank)
×
100%。利用xlfit绘制药效抑制率曲线并计算ic
50
值。细胞活性结果如表2-4所示。
[0058]
表2
[0059][0060]
表3
[0061][0062]
表4
[0063][0064]
试验例2
[0065]
靶向stat3验证
[0066]
(1)化合物112,记为dz-1,表现出良好的细胞抑制活性,采用westblot进行机制验证。hela细胞用egf(100ng/ml)处理60min,同时加入图6所示浓度的bbi-608或者dz-1处理60min,wb检测stat3的磷酸化水平,结果如图6所示。
[0067]
egf可有效促进stat3的磷酸化,为验证dz-1(化合物112)对stat3的抑制作用,在hela细胞中加入egf处理的同时加入bbi-608和dz-1,从图6可知,dz-1具有同bbi-608类似的效应,可有效降低egf激活的stat3磷酸化修饰,并且这一效果优于bbi-608。
[0068]
(2)hepg2细胞(肝癌细胞)用il-6(30ng/ml)处理60min,同时加入图7所示浓度的dz-1处理60min,wb检测stat3的磷酸化水平。
[0069]
除了egf之外,il-6也可作为激活stat3的另一重要信号分子。为验证dz-1对stat3的磷酸化抑制效果,在hepg2细胞中加入il-6处理,同时加入不同剂量的dz-1,从图7可知,dz-1(化合物112)可有效抑制il-6刺激介导的stat3磷酸化修饰,并且这一作用具有剂量依赖性。
[0070]
(3)u87细胞(神经胶质瘤细胞)用il-6(30ng/ml)处理60min,同时加入图8所示浓度的dz-1处理60min,wb检测stat3的磷酸化水平,结果如图8所示。
[0071]
从图8可知,dz-1可在人神经胶质瘤细胞u87中有效抑制il-6介导的磷酸化修饰。
[0072]
(4)ln229细胞(神经母瘤细胞)用il-6(30ng/ml)处理60min,同时加入图9所示浓度的dz-1处理60min,wb检测stat3的磷酸化水平,结果如图9所示。
[0073]
从图9可知,dz-1可在人神经母瘤细胞ln229中有效抑制il-6介导的磷酸化修饰。
[0074]
(5)huh7(肝癌细胞),ln229以及u87细胞用dz-1(0.5um)处理12h,收集细胞进行qpcr分析nanog的转录水平。
[0075]
为验证dz-1对肿瘤干细胞干性基因的影响,在huh7,ln229以及u87细胞中加入dz-1处理,随后针对stat3下游的干性基因nanog进行转录水平检测,结果见图10。从图10可知,dz-1可有效降低nanog的转录水平。
[0076]
(6)huh7,ln229,u87以及hepg2细胞用dz-1(0.5um)处理12h,收集细胞进行qpcr分析oct4的转录水平,结果见图11。从图11可知,dz-1可有效抑制stat3下游oct4基因的转录。
[0077]
(7)huh7,ln229,u87以及hepg2细胞用dz-1(0.5um)处理12h,收集细胞进行qpcr分析β-catenin的转录水平。结果见图12。从图12可知,dz-1可有效抑制stat3下游β-catenin基因的转录。
[0078]
从上述机制验证试验可知,本发明提供的化合物能够显著抑制stat3下游信号通路的转录水平,并且部分具有计量依赖性,效果强于bbi608。
[0079]
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围,其均应涵盖在本发明的权利要求和说明书的范围当中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1