一种内参、包含有该内参的试剂盒及其应用的制作方法
1.本发明属于生物技术领域,涉及一种内参、包含有该内参的试剂盒及其应用。
背景技术:
2.及时准确发现病原体对于感染性疾病的诊断和治疗意义重大。传统微生物学检验技术,诸如培养、生化鉴定、质谱等方法在厌氧、苛养微生物上存在较大局限性;而抗体/抗原,pcr等靶向检测难以广泛覆盖所有潜在的病原微生物,尤其是罕见、新发病原体。2014年,加州大学旧金山分校(ucsf)的charles chiu团队首次采用高通量测序技术(ngs,next generation sequencing)成功地对一位患有联合免疫缺陷综合征,由钩端螺旋体(leptospira)引起脑膜炎的临床患者提供病原学诊断和针对性治疗,随后,针对病原微生物的metagenomic next generation sequencing(mngs),也称为鸟枪法宏基因组测序(shotgun metagenomic sequencing)检测技术逐渐在临床广泛应用,尤其是针对免疫抑制感染患者的鉴别诊断。mngs由湿实验和干实验两个部分组成,湿实验流程一般包含微生物破壁、核酸提取、逆转录(对rna测序)、核酸片段化、文库构建、文库定量、等量混合与上机测序,干实验流程一般包含测序数据质控、人源序列过滤、微生物数据库比对、种属鉴定与丰度计算等环节。mngs具有不需培养(culture-free)、不依赖前提假设(hypothesis-free)两大优势,可以在一次检测中覆盖所有已知基因序列的病原微生物,对于疑难危重、经验性抗生素治疗无效的感染患者,以及罕见、新发病原体感染的诊断上具有很大应用价值。
3.鸟枪法宏基因组测序对临床样本中提取的总核酸进行无偏倚的鸟枪法测序,汇报微生物的序列数(reads)或根据测序数据量标化后的序列数(rpm,reads per million reads,相对于每百万条序列的数值)。因为无偏倚特性,理论上可以检测所有已知基因序列的微生物,具有常规方法所不具备的广覆盖优点。但正是由于这种无偏好性的等比例抽样(unbiased proportional sampling),最终测序获得的片段只占文库中真实核酸片段的千分之一甚至更低,因此mngs对微生物的检测性能取决于两个因素:人源核酸含量与病原核酸含量:人源核酸含量越高,微生物的检测性能越差。在人源核酸含量高的样本中,绝对载量相同的微生物的reads数或rpm会低于人源核酸含量低的样本。因此单纯通过reads数或rpm并不能反应微生物的载量高低。此外,大量病原微生物属于条件致病微生物,既可能造成感染,也可能是无症状的携带或定植。定植或感染与很多因素相关,比如微生物本身的载量(载量越高,造成感染的机率越大),微生物的毒力(毒力越高,感染几率越大),宿主的免疫状态(在免疫抑制宿主中更有可能造成感染)等等。因此,如果可以通过mngs对标本中的病原微生物进行定量检测,可以结合宿主的免疫状态对其定植或感染的状态进行更好的评估和诊断。
技术实现要素:
4.本发明的第一目的是提供一种内参,应用于宏基因组测序对微生物进行定量,通过内参核酸的测序结果反映检测的灵敏度,降低外源核酸载量对测序结果准确性的影响。
5.常规宏基因组测序对标本中所有核酸进行无差别的建库和测序,因此在某个固定的测序数据量下,病原微生物的序列数随着病原体载量,以及标本中人源核酸的含量而变化(病原微生物核酸在总核酸中的占比越高,序列数越高),无法对病原体载量进行准确评估,本发明采用通过使用特殊序列的内参相对定量,可以真实准确的反应标本中病原体核酸的载量。
6.鸟枪法mngs对微生物的检测性能受人源与病原核酸浓度的双重影响,发明人采用鸟枪法mngs对肺炎链球菌的检测性能与人源核酸含量的关系进行了试验,结果见图1,人源核酸含量越高,微生物的检测性能越差。在人源核酸含量高的样本中,绝对载量相同的微生物的reads数或rpm会低于人源核酸含量低的样本。
7.本发明提供的内参的核酸序列如seq id no.1所示,具体如下:内参的核酸序(5
’‑ꢀ3’
):aaccctatggacacaggtaacagatatagaggaaattaagctattaggaaactcaaaaacgtcatgtttgcggcgtttgcgaggcggatatttctctatgttccgcccctcacgactccgtttttacaacatacttatgacgttctaaactgagtaaacttcgctttttgattatcggaagtggggtggaaaatgtctagtaatttttaggttgatttttcgtatgacgacaattataaggttgcctacggttatgttaaccgaaactgtattttttggtcatgtcgcgtaattatttacatcccaggttacaatagatggagaataaagtaacacttaaatgactattcccaaggcttatattcgtctttcttcccttgtgagttctcacttataaccaaattattgcagttttacagccgaaattagtggaattttggagggtataggtaaatttatacttgtcgttgtttgttgtcttgttttcctttgactagttgttaagtggtatccactttgacgccttttgaacttttcgtgggacgcgtcataggtgctgtattgatactatcttcagtaagttccggacttgctacggtctacttacaagactaagtagaaatactatcggcggtttttttagccagtctcgaacaaagtgcagaagtatacatgagcagagaccaacgcaacgcgacgagagtaaaaggtagtaagtttcgagggaataaacgagtcaccaaattaactgctatgtcggcattttaagggagaaacaggcccacagtaaagttaaagttatctttgtccgcatggctgtcgctaaactggtattaacagattaagagaacatccgtatttactgttcctaagtgtacgaagtcctagtggtttacaattcttacaatttagcgaaaatggaagagaaaacaaacaagacttagggaacatttgattaagaaaaaaactaaatttctggtttagtatttggaggcag。
8.宏基因组测序对病原微生物检测的灵敏度(最低检测限)取决于文库中病原体核酸的比例,比例越高则更容易被检测。因此对某种微生物而言,载量不变的情况下,人源核酸载量越高,灵敏度越低。临床标本中人源核酸的含量受到标本类型,取样方式,患者的炎症状态等多因素影响。因为内标核酸是在精确定量后加入每一份待测的标本,因此可以通过内标核酸的测序结果反映检测灵敏度:内标序列数越低,灵敏度越低。
9.本发明提供的内参序列,与ncbi nr,genbank,refseq数据库中任意物种的序列应不存在显著相似性(blastn 0 hits)。在长度和gc含量上满足内参要求。在微生物的宏基因组测序中,可以精确反应检测灵敏度,降低测序误差。
10.本发明的第二目的是提供一种试剂盒,用于宏基因组测序对微生物进行定量,该试剂盒中包含有上述的内参。
11.本发明的第三目的是提供上述内参或者试剂盒的应用:直接应用是使用上述的内参或者试剂盒对标本中微生物进行定量。
12.最终应用是再进一步利用该定量结果,结合大数据分布统计,对标本中微生物建立定植与感染阈值,从而判定微生物定植或感染。
13.基于宏观基因组和大数据分布统计,通过微生物核酸的相对定量与不同人群(感
染人群,非感染人群)中微生物载量的分布统计,设置定植与感染的载量阈值(cutoff),为临床提供更加准确的中间结果,此阈值的建立,直接目的不是获得诊断结果或健康状况,以获取作为中间结果的信息,并不能单纯根据该建立的阈值,作为诊断患者是否患病的依据。
14.上述建立定植与感染阈值的方法,包括如下步骤:s-1. 合成如seq id no.1所示的内参核酸;s-2. pcr扩增、磁珠纯化内参核酸,使用qubit或数字pcr(ddpcr)进行定量,得到1ng内参核酸的精确分子数(摩尔数mol);s-3.在待测临床标本进行核酸提取前加入内参核酸,使得相同类型标本中的内参核酸摩尔数保持一致;s-4.对加入内参核酸的待测临床标本进行宏基因组文库构建,进行高通量测序,建库方案应使用pcr-free(片段化核酸5’端加a碱基后利用ta连接反应加上测序接头,文库构建过程中不采用pcr扩增,使得待测核酸分子的相对含量不受pcr偏好性影响),高通量测序数据量应不低于10 million reads(1000万条序列);s-5.对高通量测序结果进行比对分析,得到内参核酸每百万条序列中的测序读长数(reads per million,rpm,)rpm内参,人源序列(比对至grch38.p13)rpm宿主,以及不同病原微生物的rpm病原1,rpm病原2
……
当rpm内参 》 0时,计算index宿主 = log2(rpm宿主 / rpm内参)* 1000,index病原1 = log2(rpm病原1 / rpm内参)* 1000,index病原2 = log2(rpm病原2 / rpm内参)* 1000
……
以此类推计算得到所有检出微生物的index;s-6.采用(1)-(5)步骤所描述的方法,对同类型的一批临床标本开展宏基因组学检测。临床标本来源于两个分组:定植组与感染组。即针对某种条件致病微生物,根据其对应的临床诊疗指南选取定植组患者(微生物存在于标本中,但未引起感染症状)与感染组患者(微生物存在于标本中,且引起相应的感染症状),并统计不同分组中,某种条件致病微生物的index数值;s-7.对定植组与感染组中条件致病微生物的index数值绘制roc曲线(receiver operating characteristic curve)与pr曲线(precision recall curve),在最佳的约登指数(youden's index)时获取目标微生物定植与感染的index阈值(cut-off)。在此阈值下计算曲线下面积(area under the curve),auc必须大于等于0.8,否则需要在定植组与感染组中持续入组患者直至auc大于等于0.8。
15.通过实施上述技术方案,本发明具有如下的优点:1. 通过特定序列内参实现相对定量,可以真实准确的反应标本中微生物核酸的载量。
16.2.内参在精确定量后加入待测的标本中,可以通过内标核酸的测序结果反映检测灵敏度。
17.3.基于宏基因组的高通量测序,结合定植与感染人群的大数据统计,提供微生物定植与感染的判断标准,可以划分微生物定植与感染的载量阈值,为临床提供可供参考的中间信息。
附图说明
18.附图1为肺炎链球菌的检测性能与人源核酸含量的关系图
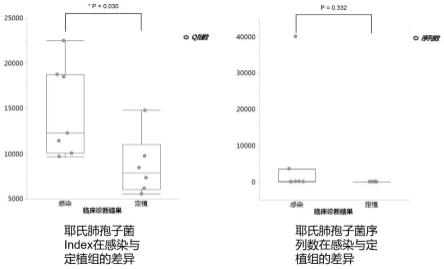
附图2为eortc/msg最新标准;附图3为耶氏肺孢子菌index和序列数在感染与定植组之间的差异;附图4为根据临床诊断分组(定植、感染)与标本检测相应的耶氏肺孢子菌的index绘制roc曲线。
具体实施方式
19.为使本发明的目的、技术方案和优点更加清楚,下面将对本发明的技术方案进行详细的描述。显然,所描述的实施例仅仅是本发明的一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动的前提下所得到的所有其它实施方式,都属于本发明所保护的范围。
实施例
20.本实施例针对耶氏肺孢子菌(pneumocystis jirovecii)设置定植与感染阈值。耶氏肺孢子菌是一种条件致病型真菌,既可以造成感染(耶氏肺孢子菌肺炎,pjp),也可能在下呼吸道中定植。本实施例中选取肺泡灌洗液(bronchoalveolar lavage fluid)进行展示,主要流程如下:(1)合成一条双链内参dna序列(5
’‑ꢀ3’
):设计原则:长度在1000
ꢀ±ꢀ
200个碱基(bp),gc含量在35-45%,其序列与ncbi nr,genbank,refseq数据库中任意物种的序列应不存在显著相似性(blastn 0 hits)具体序列如下:aaccctatggacacaggtaacagatatagaggaaattaagctattaggaaactcaaaaacgtcatgtttgcggcgtttgcgaggcggatatttctctatgttccgcccctcacgactccgtttttacaacatacttatgacgttctaaactgagtaaacttcgctttttgattatcggaagtggggtggaaaatgtctagtaatttttaggttgatttttcgtatgacgacaattataaggttgcctacggttatgttaaccgaaactgtattttttggtcatgtcgcgtaattatttacatcccaggttacaatagatggagaataaagtaacacttaaatgactattcccaaggcttatattcgtctttcttcccttgtgagttctcacttataaccaaattattgcagttttacagccgaaattagtggaattttggagggtataggtaaatttatacttgtcgttgtttgttgtcttgttttcctttgactagttgttaagtggtatccactttgacgccttttgaacttttcgtgggacgcgtcataggtgctgtattgatactatcttcagtaagttccggacttgctacggtctacttacaagactaagtagaaatactatcggcggtttttttagccagtctcgaacaaagtgcagaagtatacatgagcagagaccaacgcaacgcgacgagagtaaaaggtagtaagtttcgagggaataaacgagtcaccaaattaactgctatgtcggcattttaagggagaaacaggcccacagtaaagttaaagttatctttgtccgcatggctgtcgctaaactggtattaacagattaagagaacatccgtatttactgttcctaagtgtacgaagtcctagtggtttacaattcttacaatttagcgaaaatggaagagaaaacaaacaagacttagggaacatttgattaagaaaaaaactaaatttctggtttagtatttggaggcag。
21.(2)对内参核酸进行pcr扩增与qubit定量,质量浓度为1 ng/μl时其摩尔浓度为1.54 nm。即1 ng 内参dna的摩尔数为1.54 fmol,约有927895994个分子。
22.内参核酸序列扩增引物:正向引物(5'-3'):aaccctatggacacaggtaacag;逆向引物(5'-3'):ctgcctccaaatactaaaccag。
23.扩增程序:(3)如附图2所示,根据eortc/msg最新标准,入组临床耶氏肺孢子菌感染与定植两组患者,每组各10名患者,每位患者在纤支镜检查后取10 ml肺泡灌洗液(灌洗生理盐水体积100 ml)。
24.(4)用移液器吸取1.2 ml肺泡灌洗液转移至2ml震荡管中,使用生物样本均质仪对标本进行均质,12000 rpm离心3 min,吸取400 μl上清进行核酸,加入8 ng内标核酸、混匀后对标本进行核酸提取与pcr-free dna文库构建(酶切片段化、末端补平加a、测序接头连接、纯化)。所得文库用qpcr定量,之后对文库进行等量混合与高通量测序(illumina nextseq,se75,20m reads)。
25.(5)对定植组与感染组的标本的高通量测序结果进行分析,计算每份标本中耶氏肺孢子菌的index数值,如表1所示:表1 每份标本中耶氏肺孢子菌的index数值(6)根据临床诊断分组(定植、感染)分别统计index与序列数的差异,如图3所示,可见两组间index有显著差异(p=0.030),而序列数无显著差异(p=0.332)。
26.(7)根据临床诊断分组(定植、感染)与标本检测相应的耶氏肺孢子菌的index绘制roc曲线,如图4所示,在最佳的约登指数(youden index)取得cut-off值(index = 9769.7),可见根据此index得到的曲线下面积auc(0.881)大于0.8,因此该阈值可用作肺泡灌洗液中耶氏肺孢子菌定植与感染的区分阈值。
27.通过本实施例可以看出:1. 通过常规的测序序列数进行分析,耶氏肺孢子菌的序列数在定植与感染组患者中并无显著差异,而经过内参核酸测序结果相对定量后的index
数值,在两组间有显著差异;2.通过index数值roc分析,可以发现定植与感染患者的病原载量cut-off,从而对两组患者进行有效区分(auc=0.881),在病原体检测的基础上提供了定植与感染判断的依据,有很好的临床诊断与治疗价值。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1