一种近海沉积物自养微生物菌群及固碳途径解析方法
1.本发明涉及环境评价技术领域,尤其是涉及一种近海沉积物自养微生物菌群及固碳途径解析方法。
背景技术:
2.海草床是滨海生态系统中的重要组成部分,由于自身的生长、繁殖和死亡的过程对碳循环具有重要作用,同时还具有调节水质、捕获淡水营养盐,以及为生物提供栖息地等生态环境功能。
3.海草床面积不足海洋面积的0.2%,但为全球海洋储碳贡献高达10%,海草床生态系统巨大碳汇潜力主要来源于其相对较高的净初级生产力和缓慢的有机质分解。
4.现有技术中关于海草床固碳的研究主要是针对单一的固碳途径卡尔文循环,通过克隆文库技术等获得该循环的微生物多样性及群落结构等信息,此种方法获得微生物种类很有限,会忽略掉一些丰度相对较低的自养微生物类群。海草床沉积物是个厌氧或者缺氧的环境,在这个环境中行驶固碳功能的可能不是目前研究最普遍的卡尔文循环(主要在需氧条件下),而存在更多化能自养微生物物种及厌氧固碳途径。因此,如何解析出该环境中最主要的固碳途径,并挖掘出环境中可能丰度不高的固碳微生物,可为准确研究海草床碳循环过程以及提高海草床固碳潜力提供指导。
5.高通量测序技术已广泛应用于特定环境下微生物多样性和群落结构的研究,通过基因测序技术,获得环境中核酸和蛋白质序列,再通过将所得序列与数据库已有数据进行比对,利用相似性进行物种注释,即可以得到环境中所有物种多样性信息。
6.由于固碳自养微生物种类专一性较弱,分布较广,无法通过微生物种类来鉴定哪些是有固碳功能的自养微生物,因此,对于固碳自养生物的鉴定都是通过具有特定功能的基因(例如专门行驶固碳功能的基因)来完成,即拥有特定功能基因被作为能够行驶相应功能的微生物鉴定的分子标记。
7.作为对于特定功能基因的研究,传统的方式是通过克隆文库技术来解决,但克隆文库技术繁琐,所得数据量却相对高通量测序低得多,一个实验做几百个克隆已经耗费极大工作量。
8.因此,如何利用高通量测序技术解析近海沉积物中行驶固碳功能的微生物多样性并判断主要固碳途径,是本领域技术人员亟需解决的一项技术问题。
技术实现要素:
9.本发明的目的在于提供一种近海沉积物自养微生物菌群及固碳途径解析方法,该方法能够更加全面准确地鉴别环境样品中微生物的固碳途径及自养微生物种类。
10.本发明提供一种近海沉积物自养微生物菌群及固碳途径解析方法,包括以下步骤:
11.s1、近海沉积物样品的采集及保存;
12.s2、近海沉积物样品dna提取及质量检测;
13.s3、将质量检测合格的近海沉积物样品dna片段化,并构建pe文库;
14.s4、对近海沉积物样品进行宏基因组测序,并完成拼接组装及非冗余基因集构建;
15.s5、确定固碳途径的关键基因和蛋白,并确定其在kegg中的编号;
16.s6、通过分析并比较不同环境中不同途径的相应蛋白的基因丰度,得到特定环境中最重要的固碳途径以及不同途径的微生物种类信息;
17.其中,步骤s2和s5不限定先后顺序。
18.作为本技术方案优选地,步骤s1中,采集到的近海沉积物样品分为两组,一组置于室温条件下风干,用于理化参数的测定;另一组置于-20℃下保存,用于dna提取及宏基因组测序。
19.作为本技术方案优选地,步骤s2中,近海沉积物样品dna的质量检测包括dna浓度和纯度的检测;
20.优选地,每个样品中dna的浓度≥2ng/μl,且每个样品的量大于0.25μg。
21.作为本技术方案优选地,步骤s3具体包括:将dna片段和接头连接,筛选并去除接头自连接片段,使用pcr富集dna模板,回收pcr产物,得到pe文库;
22.优选地,所述dna片段的长度为380-420bp。
23.作为本技术方案优选地,步骤s4中,近海沉积物样品进行宏基因组测序后,进行质量控制,以去除接头和长度小于50bp、平均碱基质量值低于20的读长。
24.作为本技术方案优选地,步骤s4中,所述拼接组装时,使用拼接软件对符合质量要求的序列进行拼接组装,筛选≥300bp的重叠群,作为拼接组装结果。
25.作为本技术方案优选地,步骤s4中,所述非冗余基因集构建时,首先,对重叠群进行开放阅读框预测,选择核酸长度大于等于100bp的基因,将其翻译为氨基酸序列;对所有样品预测出来的基因序列进行聚类,每类取最长的基因作为代表序列,构建非冗余基因集。
26.作为本技术方案优选地,步骤s5中,所述固碳途径包括cbb循环、rtca循环、raccoa途径、3hp循环、3hp/4hb循环和dc/hb循环;
27.其中,cbb循环在kegg中的编号为m00165,其关键基因和蛋白为ribulose-bisphosphate carboxylase(编码基因为rbcl),蛋白在kegg中的编号为k01601;
28.rtca循环在kegg中的编号为m00173,其关键基因和蛋白为atp citrate lyase(编码基因为acla),蛋白在kegg中的编号为k15230;
29.raccoa途径在kegg中的编号为m00377,其关键基因和蛋白为5-methyltetrahydrofolate corrinoid/iron sulfur protein methyltransferase(编码基因为acse),蛋白在kegg中的编号为k15023;
30.3hp循环在kegg中的编号为m00376,其关键基因和蛋白为2-methylfumaryl-coaisomerase(编码基因为mct),蛋白在kegg中的编号为k14470;
31.3hp/4hb循环在kegg中的编号为m00375,其关键基因和蛋白为3-hydroxypropionyl-coenzyme a synthetase,蛋白在kegg中的编号为k15018;
32.dc/hb循环在kegg中的编号为m00374,其关键基因和蛋白为4-hydroxybutyrate
‑‑‑
coa ligase(编码基因为4hbl),蛋白在kegg中的编号为k14467。
33.作为本技术方案优选地,步骤s6具体包括,将非冗余基因集的氨基酸序列与kegg
数据库进行比对,获得基因对应的kegg功能;利用基因丰度获得对应功能类别的丰度,得到特定环境中的固碳途径以及不同途径的微生物种类信息,如果某样品中存在这个特征蛋白,即认为该环境中含有该条固碳途径,含有该蛋白的基因数目越多,该环境下该条途径所起的作用越大。
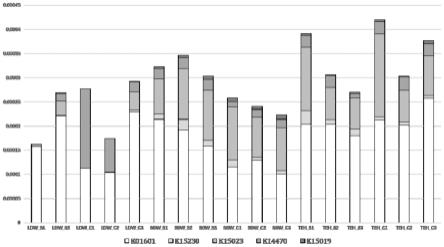
34.作为本技术方案优选地,所述理化参数包括沉积物含水量、沉积物ph、沉积物有机质含量和沉积物氮磷含量。
35.本发明的近海沉积物自养微生物菌群及固碳途径解析方法,与现有技术相比,具有以下优点:
36.本发明的近海沉积物自养微生物菌群及固碳途径解析方法主要包括:对所测样本进行宏基因组测序及物种注释,与此同时确定六条固碳途径的关键和特征蛋白以及它们在kegg数据库中的编号ko,通过不同ko丰度分析及物种注释,得到特定环境中最重要的固碳途径以及不同途径的微生物种类信息,即若在环境样本中发现了相应编码蛋白,便认为该环境存在这条固碳代谢途径,通过拥有6条不同代谢途径的微生物丰度来判断起最主要固碳作用的途径是哪条,还可解析对应途径的微生物多样性及群落结构。其与传统的克隆文库技术相比,可在同一种方法获得的数据下同时研究近海6种不同固碳途径,数据之间可以横向比较,评估不同途径在特定环境下的重要程度,而传统克隆文库技术只能单条途径去做,不同途径的重要程度就无法横向比较。并且,传统的克隆文库技术所获得的微生物多样性丰度较低,工作量很大的情况下也只能获得几百个微生物物种信息,而基于高通量测序的宏基因组手段可以获得成千上万的物种信息,通量高几个数量级,可更加全面准确地反应固碳自养微生物种群信息。
附图说明
37.为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
38.图1为本发明实施例采用位点地理位置,其中,teh代表天鹅湖,ldw代表俚岛湾,sgw代表桑沟湾;
39.图2为本发明实施例不同固碳途径中的关键蛋白丰度;
40.图3为本发明行驶cbb循环的自养生物群落结构(门水平)柱状图;
41.图4为本发明cbb循环的自养生物群落结构与环境因子相互关系的cca分析
42.图5为本发明行驶raccoa途径的自养生物群落结构(门水平)柱状图;
43.图6为本发明raccoa途径的自养生物群落结构与环境因子相互关系的rda分析;
44.图7为本发明行驶3hp循环的自养生物群落结构(纲水平)柱状图;
45.图8为本发明3hp循环的自养生物群落结构与环境因子相互关系的cca分析。
具体实施方式
46.应该指出,以下详细说明都是例示性的,旨在对本技术提供进一步的说明。除非另有指明,本文使用的所有技术和科学术语具有与本技术所属技术领域的普通技术人员通常
理解的相同含义。
47.需要注意的是,这里所使用的术语仅是为了描述具体实施方式,而非意图限制根据本技术的示例性实施方式。如在这里所使用的,除非上下文另外明确指出,否则单数形式也包括复数形式,此外,还应当理解的是,当在本说明中使用术语“包含”和/或“包括”时,其指明存在特征、步骤、操作、器件、组件和/或它们的组合。
48.下面将结合实施例对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
49.实施例
50.s1、近海沉积物样品的采集及保存
51.首先从威海3处近海代表位点进行采样,3处采样位点分别位于天鹅湖、俚岛湾和桑沟湾,每处采样位点分别在有大叶藻海草沉积物和无海草沉积物处各取3个重复,共18个样品,分别命名为teh_s1、teh_s2、teh_s3、teh_c1、teh_c2、teh_c3、ldw_s1、ldw_s2、ldw_s3、ldw_c1、ldw_c2、ldw_c3、sgw_s1、sgw_s2、sgw_s3、sgw_c1、sgw_c2、sgw_c3,后缀为字母s代表有海草的样本,后缀为字母c代表为无海草的沉积物对照。采样位点如图1所示。
52.样品采集点位于距离沉积物表面0-15cm处,采集到的近海沉积物样品分为两组,一组置于室温条件下风干,用于理化参数的测定(沉积物含水量、沉积物ph、沉积物有机质含量和沉积物氮磷含量);另一组置于-20℃下保存,用于dna提取及宏基因组测序。
53.s2、近海沉积物样品dna提取及质量检测
54.样品基因组dna提取是利用soil dna kit(omega bio-tek,美国)试剂盒,之后利用微型荧光剂tbs-380和分光光度计nanodrop200分别检测dna浓度和dna纯度性。dna样品浓度≥2ng/μl,每个样品量大于0.25μg即可进行后续pe文库的构建。
55.s3、将质量检测合格的近海沉积物样品dna片段化,并构建pe文库
56.通过自动聚焦声波基因组剪切仪(基因公司,中国)将dna片段化,筛选约400bp的片段,用于构建pe文库。
57.建库时,使用dna建库试剂盒nextflextm rapid dna-seq(bioo scientific,美国)进行建库,具体流程为先将dna片段和接头连接,然后用磁珠筛选将接头自连片段去除,并进行pcr富集dna模板,最后用磁珠回收pcr产物,得到最终pe文库。
58.s4、对近海沉积物样品进行宏基因组测序,并完成拼接组装及非冗余基因集构建
59.于上海美吉生物医药科技有限公司进行,利用illumina novaseq(illumina,美国)测序平台进行宏基因组测序。得到的数据利用fastp软件(版本0.20.0)进行质量控制,去除接头,长度小于50bp、平均碱基质量值低于20的读长;然后使用拼接软件megahit对符合质量要求的序列进行拼接组装,筛选≥300bp的重叠群(contigs)作为最终的组装结果;之后使用软件metagene对contigs进行开放阅读框(orf)预测。选择核酸长度大于等于100bp的基因,并将其翻译为氨基酸序列;用软件cd-hit对所有样品预测出来的基因序列进行聚类(参数为:90%相似度、90%覆盖度),每类取最长的基因作为代表序列,构建非冗余基因集;使用soapaligne软件分别将每个样品的高质量读长reads与非冗余基因集进行比对(95%相似度),统计基因在对应样品中的丰度信息。
60.s5、确定6种固碳途径的关键基因和蛋白,并确定其在kegg中的编号
61.cbb循环,该途径在kegg中编号为m00165,该途径关键基因和蛋白为ribulose-bisphosphate carboxylase(编码基因为rbcl),蛋白kegg编号为k01601;
62.rtca循环,该途径在kegg中编号为m00173,该途径关键基因和蛋白为atp citrate lyase(编码基因为acla),蛋白kegg编号为k15230;
63.raccoa途径,该途径在kegg中编号为m00377,该途径关键基因和蛋白为5-methyltetrahydrofolate corrinoid/iron sulfur protein methyltransferase(编码基因为acse),蛋白kegg编号为k15023;
64.3hp循环,该途径在kegg中编号为m00376,该途径关键基因和蛋白为2-methylfumaryl-coa isomerase(编码基因为mct),蛋白kegg编号为k14470;
65.3hp/4hb循环,该途径在kegg中编号为m00375,该途径关键基因和蛋白为3-hydroxypropionyl-coenzyme a dehydratase,蛋白kegg编号为k15019;
66.dc/hb循环,该途径在kegg中编号为m00374,该途径关键基因和蛋白为4-hydroxybutyrate
‑‑‑
coa ligase,(编码基因为4hbl),蛋白kegg编号为k14467。
67.s6、通过分析并比较不同环境中不同途径的相应蛋白的基因丰度,得到特定环境中最重要的固碳途径以及不同途径的微生物种类信息
68.确定6条自养生物固碳途径的关键特征蛋白在kegg数据库中的编号之后,使用软件diamond将非冗余基因集的氨基酸序列与kegg数据库进行比对(blastp比对参数设置期望值e-value为1e-5),获得基因对应的kegg功能。使用ko和module对应的基因丰度总和计算对应功能类别的丰度。
69.由图2可以看出,cbb循环为所采集环境的最主要固碳途径,第二位是raccoa固碳途径,这两种途径在桑沟湾和天鹅湖区域占绝对优势地位,其它三种固碳途径虽然在桑沟湾和天鹅湖地区也存在,但是所起作用很小;俚岛湾与二者不同,cbb循环和3hp循环是最重要的固碳途径;dc/hb循环在所有位点均没有检测到。
70.本发明进一步对沉积物样本行驶主要固碳途径的自养生物群落结构进行了分析,具体为利用各自固碳途径的关键蛋白在kegg的编号进行基因集的构建,因此构建了3个基因集,分别命名为k01601、k15023、k14470。然后对每个基因集进行物种注释,使用diamond软件将非冗余基因集的氨基酸序列与nr数据库进行比对(blastp比对参数设置期望值e-value为1e-5),并通过nr库对应的分类学信息数据库获得物种注释,然后使用物种对应的基因丰度总和计算该物种的丰度,并计算不同物种在门水平上的组成比例。并通过冗余分析rda/cca分析环境因子(氮、磷、ph、有机质等)和不同自养生物群落结构的相互关系。其中,进行cbb循环的自养生物群落结构如图3,其与环境因子的关系如图4所示:进行raccoa途径的自养生物群落结构如图5所示,其与环境因子的关系如图6所示:进行3hp循环的自养生物群落结构及如图7所示,与环境因子的关系如图8所示。
71.图3为行驶cbb循环的自养生物群落结构(门水平)柱状图,该图没有展示含量最高的变形菌门proteobacteria,proteobacteria在所有样本中丰度都最高,占绝对统治地位,最低占到70%左右,最高占到85%。从图3中可以看出,俚岛湾ldw的自养生物群落结构与桑沟湾和天鹅湖明显不同,这可能是由于俚岛湾海草床离村庄较近,受生活污染较其他两个位点严重一些。该结论同样可从图4得出(箭头代表环境因子,连线越长,代表影响越大;点
代表样本,样本点至环境因子投影点至箭头距离越小,代表影响越大),n、p含量对cbb循环的群落结构影响最显著,俚岛湾的样品明显受n、p影响较桑沟湾和天鹅湖大。俚岛湾自养生物多样性也较其他区域低,这提示有机物污染对海草床的固碳能力的破坏作用需要引起我们的重视。
72.图5为行驶raccoa途径的自养生物群落结构(门水平)柱状图,该图没有展示含量最高的变形菌门proteobacteria,proteobacteria在所有样本中丰度都最高,占绝对统治地位。从图2可知,该途径主要存在于桑沟湾和天鹅湖海草床地区,绿弯菌门chloroflexi是除了proteobacteria外丰度最高的物种。由图6可以看出,n、p和有机质含量对raccoa途径的群落结构影响最显著,有机质对桑沟湾的影响尤为显著,氮含量对天鹅湖影响最显著。
73.图7为行驶3hp循环的自养生物群落结构(纲水平)柱状图,α-proteobacteriahe在俚岛湾样本中丰度都最高,sgw和the该途径所起作用较低(图2)。由图8可以看出,n、p和ph对3hp循环的群落结构影响最显著,p对俚岛湾样品影响最大。
74.综上,通过不同固碳途径的关键蛋白在kegg数据库中的ko编号进行不同固碳途径的分析,揭示了cbb循环为威海海草床沉积物主要的固碳途径,俚岛湾由于受生活污染较严重,海草床固碳自养生物多样性降低,主要固碳途径也和桑沟湾、天鹅湖不同。因此,本发明可通过鉴定特定环境中关键和特征蛋白,间接反应该环境中的物质代谢途径,通过蛋白相对丰度的比较,明晰不同途径所起的作用重要程度;并通过对该蛋白进行物种注释,获取行驶不同途径的微生物种类的信息,由此即可完成对海草床沉积物自养微生物固碳途径及微生物群落结构的完整解析,为提高近海碳汇能力提供了理论指导。
75.最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1