一种从化合物1中分离杂质的方法及其应用与流程
1.本技术涉及杂质的分离与鉴定方法,具体涉及一种化合物1杂质的分离鉴定及其应用。
背景技术:
2.化合物1(myozone-tso),中文化学名:2-(2-((1-(3-(((1-(叔丁基)-5-氯-6-氧代-1,6-二氢哒嗪-4-基)氧)甲基)苄基)-1h-1,2,3-三唑-4-基)甲氧基)乙氧基)乙基-4-甲基苯磺酸甲酯。
3.其结构式为分子式为c
30h36
cln5o7s,分子量为646.16,是一种无色或几乎无色的黏稠液体,在甲醇、乙醇、二氯甲烷、乙腈、甲基叔丁醚和二甲基亚砜中极易溶解,在水中不溶。
4.化合物1通过糠氯酸(mca)与叔丁基肼盐酸盐反应得到二氯哒嗪,后者与1,3-苯二甲醇反应得到哒嗪衍生物,然后与tscl反应得到相应的磺酸酯,再与nan3反应得到相应的叠氮化合物,最后叠氮化合物(tcpb-n3)与pee-ots反应得到化合物1(myozone-ots)。其制备方法参考专利cn103113354a和cn107964007a。
技术实现要素:
5.目前为止没有对化合物1中含有杂质进行研究的相关资料,本技术经过大量的研究后发现化合物1在为浓缩物或在通常保存条件下时,会产生一个含量较高的杂质化合物,对化合物1的纯度有重要的影响,所述杂质的hplc相对保留时间rrt为0.70-0.81,在本技术中,其命名为杂质imp-1,分子量为1292.31,结构式如式(i)所示。
6.具体的,本技术采用如下技术方案:1. 一种如式(i)所示的化合物,
(i)。
7.2. 一种从2-(2-((1-(3-(((1-(叔丁基)-5-氯-6-氧代-1,6-二氢哒嗪-4-基)氧)甲基)苄基)-1h-1,2,3-三唑-4-基)甲氧基)乙氧基)乙基-4-甲基苯磺酸甲酯中分离如式(i)所示的化合物的方法,包括以下步骤:使用高效液相色谱法,以无机溶液和有机溶液依次为流动相对2-(2-((1-(3-(((1-(叔丁基)-5-氯-6-氧代-1,6-二氢哒嗪-4-基)氧)甲基)苄基)-1h-1,2,3-三唑-4-基)甲氧基)乙氧基)乙基-4-甲基苯磺酸甲酯进行洗脱从而分离得所述式(i)所示的化合物。
8.3. 根据项2所述的方法,所述2-(2-((1-(3-(((1-(叔丁基)-5-氯-6-氧代-1,6-二氢哒嗪-4-基)氧)甲基)苄基)-1h-1,2,3-三唑-4-基)甲氧基)乙氧基)乙基-4-甲基苯磺酸甲酯是溶解在有机溶剂中,优选所述有机溶剂选自二氯甲烷、甲基叔丁基醚、乙腈或二甲基亚砜。
9.4. 根据项2或3中任一项所述的方法,所述高效液相色谱法使用的色谱柱为十八烷基硅烷键合硅胶色谱柱。
10.5. 根据项2-4中任一项所述的方法,所述无机溶液为甲酸铵水溶液,所述有机溶液为乙腈或甲醇溶液,优选使用无机溶液和机溶液进行梯度洗脱;优选,所述甲酸铵溶液的浓度为0.02-0.05mol/l。
11.6. 根据项2-5中任一项所述的方法,其中,流动相的流速1.0ml/min。
12.7. 根据项2-6中任一项所述的方法,其中,所述高效液相色谱中检测波长为240nm。
13.8. 根据项2-7中任一项所述的方法,其中,所述高效液相色谱法中色谱柱的柱温为35℃。
14.9. 根据项2-8中任一项所述的方法,其中,所述高效液相色谱中分析时样品的进样量为5-20μl。
15.10. 式(i)的化合物在2-(2-((1-(3-(((1-(叔丁基)-5-氯-6-氧代-1,6-二氢哒嗪-4-基)氧)甲基)苄基)-1h-1,2,3-三唑-4-基)甲氧基)乙氧基)乙基-4-甲基苯磺酸甲酯的生产工艺中进行质量控制的应用,
(i)。
16.11. 根据项2-9中任一项所述的方法在2-(2-((1-(3-(((1-(叔丁基)-5-氯-6-氧代-1,6-二氢哒嗪-4-基)氧)甲基)苄基)-1h-1,2,3-三唑-4-基)甲氧基)乙氧基)乙基-4-甲基苯磺酸甲酯的生产工艺中进行质量控制的应用。
17.发明效果本技术从化合物1中分离出一个较大杂质化合物,使用本技术的分离方法,分离效果良好,并对分离出的杂质进行后处理纯化,纯化度高,并对该杂质的结构进行鉴定,成功得出了该杂质的结构,对化合物1的质量控制具有重要作用。
附图说明
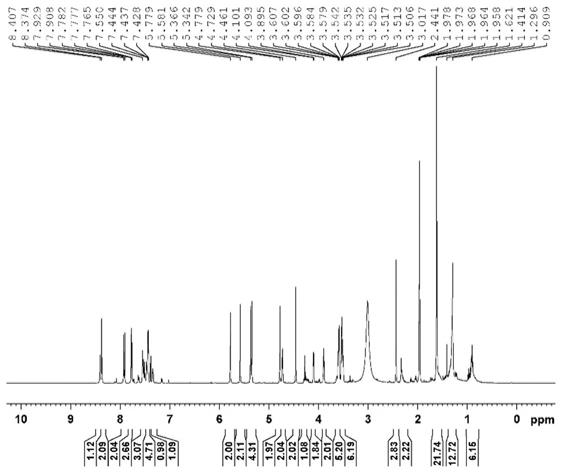
18.图1 杂质imp-1的核磁氢谱1hnmr图。
19.图2 杂质imp-1的核磁氢谱1hnmr图(重水交换)。
20.图3 杂质imp-1的核磁碳谱3cnmr图。
21.图4 杂质imp-1的lc-ms谱图。
22.图5 杂质imp-1的lc-ms质谱图。
具体实施方式
23.下面结合实施例和附图进一步说明本技术,应当理解,实施例仅用于进一步说明和阐释本技术,并非用于限制本技术。
24.除非另外定义,本说明书中有关技术的和科学的术语与本领域内的技术人员所通常理解的意思相同。虽然在实验或实际应用中可以应用与此间所述相似或相同的方法和材料,本文还是在下文中对材料和方法做了描述。在相冲突的情况下,以本说明书包括其中定义为准,另外,材料、方法和例子仅供说明,而不具限制性。以下结合具体实施例对本技术作进一步的说明,但不用来限制本技术的范围。
25.本技术提供了一种从化合物1中分离杂质化合物的方法,包括以下步骤:使用高效液相色谱法,以无机溶液和有机溶液依次为流动相对化合物1进行洗脱从而分离得所述杂质化合物。
26.色谱法(chromatography)又称“色谱分析”、“色谱分析法”、“层析法”,是一种分离和分析方法,在分析化学、有机化学、生物化学等领域有着非常广泛的应用。色谱法利用不
同物质在不同相态的选择性分配,以流动相对固定相中的混合物进行洗脱,混合物中不同的物质会以不同的速度沿固定相移动,最终达到分离的效果。
27.高效液相色谱法(high performance liquid chromatography \ hplc)又称“高压液相色谱”、“高速液相色谱”、“高分离度液相色谱”、“近代柱色谱”等。高效液相色谱是色谱法的一个重要分支,以液体为流动相,采用高压输液系统,将具有不同极性的单一溶剂或不同比例的混合溶剂、缓冲液等流动相泵入装有固定相的色谱柱,在柱内各成分被分离后,进入检测器进行检测,从而实现对试样的分析。
[0028]“外标法”与内标法相对,是指添加一定量的标准品(对照品)于空白检材中制成对照样品,与未知检材平行地进行样品处理并检测,根据对照样品响应值与其中所添加标准品(对照品)浓度的函数关系推算未知检材中被测组分浓度的定量方法。
[0029]
本技术所提供的检测方法采用高效液相色谱法高效液相色谱法的固定相有以下几种:(1)硅胶表面键合或涂渍各种聚合物;(2)其他氧化物表面涂渍聚合物;(3)无孔单分散填料;(4)有机高聚填料;(5)灌注色谱填料;(6)手性固定相填料。流动相是影响液相色谱的关键因素,高效液相色谱法中的流动相主要用水性溶剂、有机溶剂或它们的混合液。
[0030]
反相色谱填料常是以硅胶为基础,表面键合有极性相对较弱的官能团的键合相。反相色谱所使用的流动相极性较强,通常为水,缓冲液与甲醇,已腈等混合物。样品流出色谱柱的顺序是极性较强组合最先被冲出,而极性弱的组份会在色谱柱上有更强的保留。常用的反相填料有c18(ods)、c8(mos)、c4(b)、c6h5(phenyl)等。
[0031]
正相色谱用的固定相通常为硅胶(silica),以及其他具有极性官能团,如胺基团(nh2,aps)和氰基团(cn,cps)的键合相填料。由于硅胶表面的硅羟基(sioh)或其他团的极性较强,因此,分离的次序是依据样品中的各组份的极性大小,即极性强弱的组份最先被冲洗出色谱柱。正相色谱使用的流动相极性相对比固定相低,如:正乙烷(hexane),氯仿(chloroform),二氯甲烷(methylene chloride)等。
[0032]
本技术从化合物1中分离所述杂质,所述化合物1可以是任何的纯物质、浓缩物或者溶液,在一个优选的实施方式中,所述化合物1是指溶解在有机溶剂中的溶液;进一步优选,所述化合物1是溶解在二氯甲烷中的溶液;进一步优选,所述化合物1是溶解在甲基叔丁基醚的溶液;进一步优选,所述化合物1是溶解在乙腈的溶液;进一步优选,所述化合物1是溶解在二甲基亚砜中的溶液。
[0033]
本技术所提供的分离杂质化合物的方法采用的色谱柱包括但不限于上述色谱柱。在本技术优选的某些具体实施方式中,所述高效液相色谱的色谱柱为c18色谱柱。优选的,c18柱填料为十八烷基键合硅胶。
[0034]
所述无机溶液和有机溶液可以是能够实现本技术的分离目的的任何溶液,优选的,所述无机溶液为甲酸铵水溶液,浓度优选为0.02-0.05mol/l,例如所述甲酸铵水溶液的浓度可以为0.02mol/l、025mol/l、0.03mol/l、0.035mol/l、0.04mol/l、0.045mol/l、0.5mol/l,所述有机溶液为乙腈或甲醇溶液。
[0035]
进一步优选的,本技术采用梯度洗脱的方式进行分离。
[0036]
所述梯度洗脱(gradient elution)又称为梯度淋洗或程序洗脱。在同一个分析周
期中,按一定程度不断改变流动相的浓度配比,称为梯度洗脱。在气相色谱中,为了改善对宽沸程样品的分离和缩短分析周期,广泛采用程序升温的方法。而在液相色谱中对组分复杂的样品则采用梯度洗脱的方法。在同一个分析周期中,按一定程序不断改变流动相的浓度配比,称为梯度洗脱。从而可以使一个复杂样品中的性质差异较大的组分能按各自适宜的容量因子k达到良好的分离目的。
[0037]
在本技术一个优选的实施方式中,所述流动相的流速为1.0ml/min。
[0038]
在本技术一个优选的实施方式中,所述高效液相色谱中检测波长为240nm。
[0039]
在本技术一个优选的实施方式中,所述高效液相色谱法中色谱柱的柱温为35℃。
[0040]
在本技术一个优选的实施方式中,所述高效液相色谱中分析时样品的进样量为5-20μl,例如可以为5μl、6μl、7μl、8μl、9μl、10μl、11μl、12μl、13μl、14μl、15μl、16μl、17μl、18μl、19μl、20μl。
[0041]
本技术中所分离出的杂质化合物经过鉴定,其结构式如(i)所示,(i)。
[0042]
本技术进一步提供了一种所分离的杂质化合物在化合物1质量控制中的应用,例如产品纯度检测、质量控制。通过该杂质的结构的确定,化合物1产生该杂质的机理就可以确定下来:产生的主要路径是双分子的偶联,通过理论分析,降低化合物的浓度和保存温度,即可有效降低分子间的碰撞,从而达到控制化合物质量的目的。
实施例
[0043]
下面通过具体的制备实施例进一步说明本技术。
[0044]
1、仪器及试剂液相色谱仪(agilent,1260)、分析天平(sartorius,cpa225d)、制备型hplc(北京创新通恒科技有限公司,lc3000i)化合物1(北京师范大学化学学院)、乙腈(thermo fisher,hplc级)、甲酸铵(天津市光复精细化工研究所,hplc级)2、色谱条件色谱柱:waters sunfire
tm prep c18 (10
×
250mm 5μm);流动相 a:乙腈(acn);流动相b:20mmol/l甲酸铵溶液,配置过程:称取1.26g甲酸铵,溶于1000ml纯化水
中混匀,过滤超声即得;流速:1.0 ml/min;柱温:35℃;检测波长:240 nm;进样量:5-20μl;梯度洗脱表如表1所示,表1 hplc梯度表3、溶液配制将0.2ml样品存放于1ml液相小瓶中,向此小瓶中加入约0.6ml acn,混匀。共配置化合物1-杂质分离-1、化合物1-杂质分离-2、化合物1-杂质分离-3三个批次。
[0045]
4、测定分别取化合物1-杂质分离-1、化合物1-杂质分离-2、化合物1-杂质分离-3的溶液进样,按照上述色谱条件进行检测。
[0046]
5、结果杂质imp-1和化合物1分离效果很好,平均保留时间和相对保留时间如下表2。
[0047]
表2 保留时间表制备hplc的纯化效果情况如表3表3 纯化数据表由表3可以看出,样品在当前制备hplc条件下,分离纯化效果较好。对制备出来的馏分经过萃取旋蒸,即可得到目标化合物,即杂质imp-1。
[0048]
采用在上述条件进行hplc分离后,对得到的杂质溶液即imp-1使用乙酸乙酯(ea)进行萃取以去除盐和水。考虑到溶剂的毒性,挥发性和稳定性等因素,本技术中使用乙酸乙酯(ea)作为萃取剂。
[0049]
具体操作为,对分离后的杂质溶液进行三次ea萃取,每次萃取的相体积比约为1:1,合并流分后在30℃条件下旋蒸,短时间干燥样品即可得干燥后的纯品。对于最终少量残存的水,可配合使用冻干手段。
[0050]
6. 杂质鉴定将样品1所分离纯化后的杂质imp-1纯品进行核磁检测,所用仪器信息和溶剂如下所示,仪器型号和厂家:avance iii 500 bruker a&t center bnu;溶剂:cd3cn。
[0051]
得到核磁氢谱图和碳谱图分别见图1、2、3,核磁图谱解析见表4和表5。
[0052]
表4 氢谱解析表5 碳谱解析
ꢀ
结合化合物1结构以及核磁和分子量的结果,推测杂质imp-1(rrt0.70-0.81)为化合物1分子的亲电性基团(对甲苯磺酸酯)和亲核性集团(三氮唑)发生分子间反应的产物,即1-(3-(((1-(叔-丁基)-5-氯-6-氧代-1,6-二氢哒嗪-4-基)氧)甲基)苄基)-3-(2-(2-((1-(3-(((1-(叔-丁基)-5-氯-6-氧代-1,6-二氢哒嗪-4-基)氧)甲基)苄基)-1h-1,2,3-三唑-4-基)甲氧基)乙氧基)乙基)-4-((2-(2-(甲基苯磺酰)乙氧基)乙氧基)甲基)-1h-1,2,3-三唑-3-正离子-4-甲基苯磺酸甲酯,结构式如(i)所示,分子式为c
60h72
cl2n
10o14
s2,分子量1292.31,阴离子为对甲基苯磺酸,即式(i)所示的化合物,因核磁检测用杂质样品为化合物1经制备柱分离纯化得到,阴离子发生交换,所以核磁氢谱和碳谱解析数据中无甲基苯磺酸的信号。
[0053]
(i)所述杂质imp-1的理论分子量为1291.31,减去阴离子对甲基苯磺酸根的分子量172.2016,即1120.1084,与样品1所分离纯化出的杂质imp-1纯品的实测结果基本一致,见图4和图5。本技术的鉴定结果准确,得到了杂质imp-1的结构式。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1