一种取代苯甲酸二十二碳六烯酰胺乙基酯衍生物及其制备方法与应用与流程
1.本发明属于小分子活性化合物技术领域,具体而言,涉及一种化合物及其系列衍生物,尤其是涉及一种取代苯甲酸二十二碳六烯酰胺乙基酯衍生物及其制备方法与应用。
背景技术:
2.临床研究表明,饮食中富含的ω-3脂肪酸(ω-3fas)二十二碳六烯酸(dha)和二十碳五烯酸(epa)有益于心血管疾病、神经系统疾病和炎症相关疾病,对血管炎症,神经炎症,高血压和血栓形成提供保护。越来越多的实验证据表明,这些作用是通过氧化和非氧化的介导作用将ω-3fas转化为具有生物活性的脂质代谢产物。
3.不饱和脂肪酰乙醇胺是一类内源性脂质代谢产物,广泛存在于大脑、肠道和血液中。其中的二十二碳六烯酰乙醇胺(dhea)和二十二碳五烯酰乙醇胺(dpea)可以剂量依赖地抑制lps诱导的巨噬细胞raw264.7促炎因子的增加,并抑制raw 264.7中的no的生成。表面等离子共振(spr)测试表明dhea和dpea都能与nur77具有中等结合亲和力。分子对接技术也证实dhea和dpea是nur77的配体。此外,dhea和dpea可显著降低lps-诱导炎症,并且是通过nur77依赖性方式来阻断nf-κb活性的。
4.虽然研究表明现有ω-3多不饱和脂肪酸具有增强免疫和抗炎作用,但是这些天然的多不饱和脂肪酸及其内源性脂质代谢产物不饱和脂肪酰乙醇胺的抗炎活性不太理想,因需要对其进行结构修饰以提高生物活性,从而可应用于医药领域。
技术实现要素:
5.鉴于现有技术的不足,本发明的目的在于提供一种取代苯甲酸二十二碳六烯酰胺乙基酯衍生物及其制备方法,以期从中筛选潜在的可用于开发具有抗炎活性的海洋脂类活性药物。
6.为了实现上述技术目的,本发明人创造性地在不饱和脂肪酰乙醇胺的羟基位引进了取代苯甲酸的分子片段,合成了一系列取代苯甲酸二十二碳六烯酰胺乙基酯衍生物,从而有效增强了不饱和脂肪酰乙醇胺化合物的抗炎活性。
7.具体地,实现本发明技术目的的技术方案概括如下:一种取代苯甲酸二十二碳六烯酰胺乙基酯衍生物,该衍生物的结构通式如下:
[0008][0009]
其中,r1=h,cl,ch3,cf3,cn或ch2ph;r2=h,f,cl,br,i,ch3,ch2ch2ch2ch3,och3,sch3或och2ch3。
[0010]
进一步优选地,如上所述的取代苯甲酸二十二碳六烯酰胺乙基酯衍生物,包括苯甲酸-(二十二碳六烯酰胺乙基)-酯(j1),4-氟苯甲酸-(二十二碳六烯酰胺乙基)-酯(j2),
4-氯苯甲酸-(二十二碳六烯酰胺乙基)-酯(j3),4-溴苯甲酸-(二十二碳六烯酰胺乙基)-酯(j4),4-碘苯甲酸-(二十二碳六烯酰胺乙基)-酯(j5),4-甲基苯甲酸-(二十二碳六烯酰胺乙基)-酯(j6),4-正丁基苯甲酸-(二十二碳六烯酰胺乙基)-酯(j7),4-甲氧基苯甲酸-(二十二碳六烯酰胺乙基)-酯(j8),4-巯基苯甲酸-(二十二碳六烯酰胺乙基)-酯(j9),4-乙氧基苯甲酸-(二十二碳六烯酰胺乙基)-酯(j10),3-苄基苯甲酸-(二十二碳六烯酰胺乙基)-酯(j11),2-三氟甲基苯甲酸-(二十二碳六烯酰胺乙基)-酯(j12),3-腈基苯甲酸-(二十二碳六烯酰胺乙基)-酯(j13),2-甲基-5-氟苯甲酸-(二十二碳六烯酰胺乙基)-酯(j14),2,4-二氯苯甲酸-(二十二碳六烯酰胺乙基)-酯(j15),2,6-二氯苯甲酸-(二十二碳六烯酰胺乙基)-酯(j16),3-甲基-4-氟苯甲酸-(二十二碳六烯酰胺乙基)-酯(j17),正十六烷酸-(二十二碳六烯酰胺乙基)-酯(j18)等十八种。
[0011]
另外,本发明将二十二碳六烯酰乙醇胺和取代苯甲酰氯在缚酸剂的作用下,制备得到系列取代苯甲酸二十二碳六烯酰胺乙基酯衍生物。因此,本发明还提供了一种上述取代苯甲酸二十二碳六烯酰胺乙基酯衍生物的制备方法,该方法包括以下步骤:
[0012]
s1:将富含二十二碳六烯酸的油脂和乙醇胺混合,在不添加任何溶剂的情况下,于75-85℃加热搅拌反应10-20h,反应结束后,硅胶拌样,以v
乙酸乙酯
∶v
石油醚
=(1.8-2.2)∶1作为洗脱液快速进行硅胶柱层析纯化,然后采用高效液相制备色谱进一步纯化,收集保留时间t=19.20-21.80min的组分,浓缩即得淡黄色粘稠状产物二十二碳六烯酰乙醇胺;
[0013]
s2:取s1步骤制得的二十二碳六烯酰乙醇胺,与取代苯甲酰氯均匀混合溶于有机溶剂中,在缚酸剂的作用下,于0-5℃下搅拌反应8-24h,反应结束后,1,2-二氯乙烷萃取1-4次,无水mgso4干燥,浓缩,以v
乙酸乙酯
∶v
石油醚
=(0.8-1.2)∶1作为洗脱液快速进行硅胶柱层析纯化,然后采用高效液相制备色谱进一步纯化,收集特征保留时间t
1/2
=8.04-20.71min的组分,浓缩即得淡黄色粘稠状取代苯甲酸二十二碳六烯酰胺乙基酯衍生物。
[0014]
进一步优选地,如上所述的取代苯甲酸二十二碳六烯酰胺乙基酯衍生物的制备方法,步骤s1中所述富含二十二碳六烯酸的油脂选自藻油、鱼油、海狗油和磷虾油中的一种或两种以上。
[0015]
进一步优选地,如上所述的取代苯甲酸二十二碳六烯酰胺乙基酯衍生物的制备方法,步骤s2中所述取代苯甲酰氯选自如下的一种或两种以上:苯甲酰氯,4-氟苯甲酰氯,4-氯苯甲酰氯,4-溴苯甲酰氯,4-碘苯甲酰氯,4-甲基苯甲酰氯,4-正丁基苯甲酰氯,4-甲氧基苯甲酰氯,4-巯基苯甲酰氯,4-乙氧基苯甲酰氯,3-苄基苯甲酰氯,2-三氟甲基苯甲酰氯,3-腈基苯甲酰氯,2-甲基-5-氟苯甲酰氯,2,4-二氯苯甲酰氯,2,6-二氯苯甲酰氯,3-甲基-4-氟苯甲酰氯,正十六烷酰氯。
[0016]
进一步优选地,如上所述的取代苯甲酸二十二碳六烯酰胺乙基酯衍生物的制备方法,步骤s2中所述的有机溶剂选自如下的一种:四氢呋喃,二氯甲烷,1,2-二氯乙烷,二氧六环,乙腈。
[0017]
进一步优选地,如上所述的取代苯甲酸二十二碳六烯酰胺乙基酯衍生物的制备方法,步骤s2中所述的缚酸剂选自如下的一种:三乙胺(et3n),二乙胺(dea),六氢吡啶(pd)。
[0018]
进一步优选地,如上所述的取代苯甲酸二十二碳六烯酰胺乙基酯衍生物的制备方法,步骤s2中所述搅拌反应8-24h,是指反应时间为8h,12h,16h,20h和24h中的一个时间点。
[0019]
另外,本发明还提供了一种上述取代苯甲酸二十二碳六烯酰胺乙基酯衍生物在制
备抗炎药物中的应用。具体地,所述的抗炎药物能下调机体的促炎性介质tnf-α,il-6和il-1β,以及下调一氧化氮的外分泌水平。
[0020]
与现有技术相比,本发明制备的取代苯甲酸二十二碳六烯酰胺乙基酯衍生物是一种结构新颖的化合物,这些化合物具有显著抑制raw264.7巨噬细胞中促炎因子的功效。采用mtt法检测(j1-j18)系列化合物的细胞毒性,发现化合物浓度为10μm时,在小鼠巨噬细胞raw264.7、人大细胞肺癌细胞h460、人正常肝细胞lo2中没有明显的细胞毒性。该系列取代苯甲酸二十二碳六烯酰胺乙基酯衍生物对raw264.7中由脂多糖lps诱导的促炎性介质(tnf-α,il-6和il-1β)的mrna表达具有一定的下调作用,以及能抑制raw264.7细胞中no的产生,同时能够抑制促炎性细胞因子(tnf-α和il-1β)的蛋白表达。因此,本发明提供的取代苯甲酸二十二碳六烯酰胺乙基酯衍生物是一类具备抗炎生物活性的新型海洋脂类药物,具有生物利用率高,安全性好等优点,具有被开发成海洋脂类抗炎小分子药物的潜力。
附图说明
[0021]
图1:化合物j1的1h nmr氢谱图。
[0022]
图2:化合物j1的
13
c nmr碳谱图。
[0023]
图3:取代苯甲酸二十二碳六烯酰胺乙基酯衍生物(j1-j18)的合成路线图。
[0024]
图4:化合物j1-j18对raw264.7细胞24小时存活率的影响。
[0025]
图5:化合物j1-j18对h460细胞24小时存活率的影响。
[0026]
图6:化合物j1-j18对lo2细胞24小时存活率的影响。
[0027]
图7:化合物j1-j18抑制raw264.7细胞中no的产生。
[0028]
图8:化合物j1-j18下调促炎介质tnf-α的mrna表达。
[0029]
图9:化合物j1-j18下调促炎介质il-6的mrna表达。
[0030]
图10:化合物j1-j18下调促炎介质il-1β的mrna表达。
[0031]
图11:化合物j1-j18抑制促炎性细胞因子il-1β和tnf-α的蛋白表达。
具体实施方式
[0032]
本发明提供了取代苯甲酸二十二碳六烯酰胺乙基酯衍生物(j1-j18)的合成路线图,参见图3。上述化合物的制备方法,包括如下步骤:
[0033]
s1:将10g富含二十二碳六烯酸的油脂和20g的乙醇胺混合,在不添加任何溶剂的情况下,80℃加热搅拌反应15h,反应结束后,硅胶拌样,快速硅胶柱层析纯化(v
乙酸乙酯
∶v
石油醚
=2∶1)和高效液相制备色谱进一步纯化,收集特征保留时间t
1/2
=20.56min的组分,浓缩即得淡黄色粘稠状产物二十二碳六烯酰乙醇胺。
[0034]
s2:将1.0mmol的二十二碳六烯酰乙醇胺和1.1mmol的取代苯甲酰氯均匀混合溶于有机溶剂中,在缚酸剂的作用下,0-5℃下搅拌反应8-24h,反应结束后,1,2-二氯乙烷萃取两次,无水mgso4干燥,浓缩,快速硅胶柱层析纯化(v
乙酸乙酯
∶v
石油醚
=1∶1)和高效液相制备色谱进一步纯化,收集特征保留时间t
1/2
=8.04-20.71min的组分,浓缩即得淡黄色粘稠状取代苯甲酸二十二碳六烯酰胺乙基酯衍生物(j1-j18)。
[0035]
为了使本领域技术人员更好地理解本发明的技术方案并能予以实施,下面结合具体实施例对本发明作进一步说明,但所举实施例不作为对本发明保护范围的限定。
[0036]
实施例1:化合物苯甲酸-(二十二碳六烯酰胺乙基)-酯(j1)的制备
[0037]
化合物(j1)的结构式为:
[0038][0039]
将1.0mmol的二十二碳六烯酰乙醇胺和1.1mmol的苯甲酰氯均匀混合溶于1,2-二氯乙烷中,在缚酸剂et3n的作用下,0-5℃下搅拌反应8h,反应结束后,1,2-二氯乙烷萃取两次,无水mgso4干燥,浓缩,快速硅胶柱层析纯化(v
乙酸乙酯
∶v
石油醚
=1∶1)和高效液相制备色谱进一步纯化,收集特征保留时间t
1/2
=9.78min的组分,浓缩即得淡黄色粘稠状产物j1。
[0040]
化合物(j1)的1h nmr氢谱图如图1所示,
13
c nmr碳谱图如图2所示,具体的氢谱和碳谱数据如下:
[0041]
淡黄色粘稠状物,1h nmr(400mhz,cdcl3,ppm):δ8.07-8.05(m,2h,arh),7.62-7.58(m,1h,arh),7.49-7.46(m,2h,arh),5.90(s,1h,ch=ch),5.45-5.32(m,11h,ch=ch),4.45-4.43(m,2h,nhch2),3.70-3.66(m,2h,och2),2.87-2.83(m,8h,4ch2),2.45-2.40(m,2h,ch2),2.29-2.25(m,2h,ch2),2.11-2.06(m,2h,ch2),1.72(br,1h,nh),1.32-1.27(m,2h,ch2),1.01-0.97(m,3h,ch3);
13
c nmr(100mhz,cdcl3,ppm):δ172.6,166.7(c=o),133.3,132.1,130.2,130.1,129.8,129.7,128.6,128.5,128.3,128.2,128.1,128.0,127.9,127.0,63.8(ch2o),38.9(ch2nh),36.8,31.5,29.3,29.1,27.2,25.6,23.3,20.6,14.3(ch3).hrms(tof-ms,+):m/z[m+na]
+
calculated for c
31h41
nnao
3+
498.2979,found 498.2968。
[0042]
实施例2:化合物4-氟苯甲酸-(二十二碳六烯酰胺乙基)-酯(j2)的制备
[0043]
化合物(j2)的结构式为:
[0044][0045]
将1.0mmol的二十二碳六烯酰乙醇胺和1.1mmol的4-氟苯甲酰氯均匀混合溶于四氢呋喃中,在缚酸剂et3n的作用下,0-5℃下搅拌反应12h,反应结束后,1,2-二氯乙烷萃取两次,无水mgso4干燥,浓缩,快速硅胶柱层析纯化(v
乙酸乙酯
∶v
石油醚
=1∶1)和高效液相制备色谱进一步纯化,收集特征保留时间t
1/2
=9.87min的组分,浓缩即得淡黄色粘稠状产物j2。
[0046]
化合物(j2)的主要氢谱和碳谱数据如下:
[0047]
淡黄色粘稠状物,1h nmr(400mhz,cdcl3,ppm):δ8.09-8.05(m,2h,arh),7.16-7.12(m,2h,arh),5.88(s,1h,ch=ch),5.42-5.31(m,11h,ch=ch),4.44-4.41(m,2h,nhch2),3.69-3.65(m,2h,och2),2.87-2.83(m,8h,4ch2),2.45-2.40(m,2h,ch2),2.29-2.25(m,2h,ch2),2.13-2.05(m,2h,ch2),1.73(br,1h,nh),1.33-1.27(m,2h,ch2),1.01-0.97(m,3h,ch3);
13
c nmr(100mhz,cdcl3,ppm):δ172.6,167.2(c=o),165.8,132.3,132.2,132.1,129.4,128.6,128.4,128.3,128.2,128.1,128.0,127.9,127.0,126.0,115.8,115.6,63.9(ch2o),38.9(ch2nh),36.4,31.3,30.2,29.4,27.8,25.7,23.3,20.6,14.3(ch3).hrms(tof-ms,+):m/z[m+na]
+
calculated for c
31h40
fnnao
3+
516.2884,found 516.2863。
[0048]
实施例3:化合物4-氯苯甲酸-(二十二碳六烯酰胺乙基)-酯(j3)的制备
[0049]
化合物(j3)的结构式为:
[0050][0051]
将1.0mmol的二十二碳六烯酰乙醇胺和1.1mmol的4-氯苯甲酰氯均匀混合溶于1,2-二氯乙烷中,在缚酸剂et3n的作用下,0-5℃下搅拌反应16h,反应结束后,1,2-二氯乙烷萃取两次,无水mgso4干燥,浓缩,快速硅胶柱层析纯化(v
乙酸乙酯
∶v
石油醚
=1∶1)和高效液相制备色谱进一步纯化,收集特征保留时间t
1/2
=11.40min的组分,浓缩即得淡黄色粘稠状产物j3。
[0052]
化合物(j3)的主要氢谱和碳谱数据如下:
[0053]
淡黄色粘稠状物,1h nmr(400mhz,dmso-d6,ppm):δ7.99-7.97(m,2h,arh),7.45-7.43(m,2h,arh),5.95(s,1h,ch=ch),5.55-5.31(m,11h,ch=ch),4.44-4.41(m,2h,nhch2),3.69-3.65(m,2h,och2),2.87-2.83(m,8h,4ch2),2.43-2.39(m,2h,ch2),2.28-2.25(m,2h,ch2),2.10-2.05(m,2h,ch2),1.85(br,1h,nh),1.41-1.20(m,2h,ch2),1.00-0.96(m,3h,ch3);
13
c nmr(100mhz,cdcl3,ppm):δ172.6,165.9(c=o),139.8,132.1,131.1,130.2,130.0,129.5,128.8,128.6,128.3,128.2,128.1,128.0,127.9,127.0,64.3(ch2o),38.0(ch2nh),35.6,31.4,29.2,27.2,25.6,23.3,22.6,20.6,14.3(ch3).hrms(tof-ms,+):m/z[m+na]
+
calculated for c
31h40
clnnao
3+
532.2589,found 532.2583。
[0054]
实施例4:化合物4-溴苯甲酸-(二十二碳六烯酰胺乙基)-酯(j4)的制备
[0055]
化合物(j4)的结构式为:
[0056][0057]
将1.0mmol的二十二碳六烯酰乙醇胺和1.1mmol的4-溴苯甲酰氯均匀混合溶于二氯甲烷中,在缚酸剂dea的作用下,0-5℃下搅拌反应20h,反应结束后,1,2-二氯乙烷萃取两次,无水mgso4干燥,浓缩,快速硅胶柱层析纯化(v
乙酸乙酯
∶v
石油醚
=1∶1)和高效液相制备色谱进一步纯化,收集特征保留时间t
1/2
=12.38min的组分,浓缩即得淡黄色粘稠状产物j4。
[0058]
化合物(j4)的主要氢谱和碳谱数据如下:
[0059]
淡黄色粘稠状物,1h nmr(400mhz,cdcl3,ppm):δ7.91-7.89(m,2h,arh),7.61-7.59(m,2h,arh),5.91(s,1h,ch=ch),5.41-5.38(m,11h,ch=ch),4.43-4.41(m,2h,nhch2),3.67-3.65(m,2h,och2),2.88-2.83(m,10h,5ch2),2.42-2.40(m,2h,ch2),2.28-2.25(m,2h,ch2),2.10-2.07(m,2h,ch2),1.27(br,1h,nh),1.00-0.96(m,3h,ch3);
13
c nmr(100mhz,cdcl3,ppm):δ172.7,166.0,(c=o),132.1,131.8,131.2,129.5,128.7,128.6,128.5,128.4,128.3,128.1,128.0,127.9,127.0,64.0(ch2o),38.8(ch2nh),36.4,29.7,25.7,25.6,25.5,23.3,20.6,14.3(ch3).hrms(tof-ms,+):m/z[m+na]
+
calculated for c
31h40
brnnao
3+
576.2084,found 576.2056。
[0060]
实施例5:化合物4-碘苯甲酸-(二十二碳六烯酰胺乙基)-酯(j5)的制备
[0061]
化合物(j5)的结构式为:
[0062][0063]
将1.0mmol的二十二碳六烯酰乙醇胺和1.1mmol的4-碘苯甲酰氯均匀混合溶于二氧六环中,在缚酸剂pd的作用下,0-5℃下搅拌反应24h,反应结束后,1,2-二氯乙烷萃取两
次,无水mgso4干燥,浓缩,快速硅胶柱层析纯化(v
乙酸乙酯
∶v
石油醚
=1∶1)和高效液相制备色谱进一步纯化,收集特征保留时间t
1/2
=12.24min的组分,浓缩即得淡黄色粘稠状产物j5。
[0064]
化合物(j5)的主要氢谱和碳谱数据如下:
[0065]
淡黄色粘稠状物,1h nmr(400mhz,cdcl3,ppm):7.84-7.82(m,2h,arh),7.76-7.74(m,2h,arh),5.90-5.80(s,1h,ch=ch),5.43-5.31(m,11h,ch=ch),4.43-4.41(m,2h,nhch2),3.67-3.66(m,2h,och2),2.85-2.83(m,8h,4ch2),2.41-2.39(m,2h,ch2),2.28-2.18(m,2h,ch2),2.11-2.07(m,2h,ch2),1.78(br,1h,nh),1.28-1.27(m,2h,ch2),1.01-0.96(m,3h,ch3);
13
c nmr(100mhz,cdcl3,ppm):172.6,166.2(c=o),137.9,132.1,131.1,130.2,130.0,129.5,129.2,128.6,128.4,128.3,128.1,128.0,127.9,127.0,64.0(ch2o),38.8(ch2nh),36.7,36.4,29.3,27.2,25.6,23.3,22.6,20.6,14.3(ch3).hrms(tof-ms,+):m/z[m+na]
+
calculated for c
31h40
innao
3+
624.1945,found 624.1924。
[0066]
实施例6:化合物4-甲基苯甲酸-(二十二碳六烯酰胺乙基)-酯(j6)的制备
[0067]
化合物(j6)的结构式为:
[0068][0069]
将1.0mmol的二十二碳六烯酰乙醇胺和1.1mmol的4-甲基苯甲酰氯均匀混合溶于乙腈中,在缚酸剂et3n的作用下,0-5℃下搅拌反应8h,反应结束后,1,2-二氯乙烷萃取两次,无水mgso4干燥,浓缩,快速硅胶柱层析纯化(v
乙酸乙酯
∶v
石油醚
=1∶1)和高效液相制备色谱进一步纯化,收集特征保留时间t
1/2
=10.60min的组分,浓缩即得淡黄色粘稠状产物j6。
[0070]
化合物(j6)的主要氢谱和碳谱数据如下:
[0071]
淡黄色粘稠状物,1h nmr(400mhz,cdcl3,ppm):7.95-7.93(m,2h,arh),7.28-7.26(m,2h,arh),5.94(s,1h,ch=ch),5.44-5.31(m,11h,ch=ch),4.43-4.40(m,2h,nhch2),3.69-3.65(m,2h,och2),2.87-2.83(m,8h,4ch2),2.43(s,3h,ch3),2.42-2.40(m,2h,ch2),2.29-2.25(m,2h,ch2),2.11-2.05(m,2h,ch2),1.76(br,1h,nh),1.31-1.27(m,2h,ch2),1.01-0.96(m,3h,ch3);
13
c nmr(100mhz,cdcl3,ppm):172.6,166.8(c=o),144.0,132.1,129.7,129.4,129.2,128.6,129.3,128.2,128.1,128.0,128.0,127.9,127.1,127.0,63.6(ch2o),39.0(ch2nh),36.4,31.5,29.4,27.2,25.6,25.5,23.3,21.7,14.3(ch3),14.1.hrms(tof-ms,+):m/z[m+na]
+
calculated for c
32h43
nnao
3+
512.3135,found 512.3129。
[0072]
实施例7:化合物4-正丁基苯甲酸-(二十二碳六烯酰胺乙基)-酯(j7)的制备
[0073]
化合物(j7)的结构式为:
[0074][0075]
将1.0mmol的二十二碳六烯酰乙醇胺和1.1mmol的4-正丁基苯甲酰氯均匀混合溶于二氯甲烷中,在缚酸剂pd的作用下,0-5℃下搅拌反应12h,反应结束后,1,2-二氯乙烷萃取两次,无水mgso4干燥,浓缩,快速硅胶柱层析纯化(v
乙酸乙酯
∶v
石油醚
=1∶1)和高效液相制备色谱进一步纯化,收集特征保留时间t
1/2
=15.93min的组分,浓缩即得淡黄色粘稠状产物j7。
[0076]
化合物(j7)的主要氢谱和碳谱数据如下:
[0077]
淡黄色粘稠状物,1h nmr(400mhz,cdcl3,ppm):7.95-7.93(m,2h,arh),7.26-7.23(m,2h,arh),6.14(s,1h,ch=ch),5.38-5.30(m,11h,ch=ch),4.41-4.38(m,2h,nhch2),
3.66-3.62(m,2h,och2),2.86-2.82(m,8h,4ch2),2.69-2.65(m,2h,ch2),2.42-2.39(m,2h,ch2),2.27-2.24(m,2h,ch2),2.10-2.05(m,2h,ch2),1.63-1.58(m,2h,ch2),1.38-1.28(m,4h,2ch2),1.26(br,1h,nh),0.99-0.96(m,3h,ch3),0.93-0.89(m,3h,ch3);
13
c nmr(100mhz,cdcl3,ppm):172.6,166.8(c=o),148.9,132.0,129.7,129.3,128.6,128.5,128.3,128.2,128.1,128.1,128.0,128.0,127.9,127.2,127.0,63.6(ch2o),39.0(ch2nh),36.4,35.7,33.3,32.831.8,28.7,26.0,25.6,23.4,22.3,20.6,14.3(ch3),13.9.hrms(tof-ms,+):m/z[m+na]
+
calculated for c
35h49
nnao
3+
554.3605,found 554.3593。
[0078]
实施例8:化合物4-甲氧基苯甲酸-(二十二碳六烯酰胺乙基)-酯(j8)的制备
[0079]
化合物(j8)的结构式为:
[0080][0081]
将1.0mmol的二十二碳六烯酰乙醇胺和1.1mmol的4-甲氧苯甲酰氯均匀混合溶于1,2-二氯乙烷中,在缚酸剂dea的作用下,0-5℃下搅拌反应16h,反应结束后,1,2-二氯乙烷萃取两次,无水mgso4干燥,浓缩,快速硅胶柱层析纯化(v
乙酸乙酯
∶v
石油醚
=1∶1)和高效液相制备色谱进一步纯化,收集特征保留时间t
1/2
=9.65min的组分,浓缩即得淡黄色粘稠状产物j8。
[0082]
化合物(j8)的主要氢谱和碳谱数据如下:
[0083]
淡黄色粘稠状物,1h nmr(400mhz,cdcl3,ppm):8.01-7.99(m,2h,arh),6.95-6.93(m,2h,arh),5.93(s,1h,ch=ch),5.39-5.35(m,11h,ch=ch),4.42-4.39(m,2h,nhch2),3.89(s,3h,och3),3.66-3.65(m,2h,och2),2.86-2.84(m,8h,4ch2),2.43-2.38(m,2h,ch2),2.28-2.20(m,2h,ch2),2.10-2.05(m,2h,ch2),1.75(br,1h,nh),1.32-1.27(m,2h,ch2),1.00-0.97(m,3h,ch3);
13
c nmr(100mhz,cdcl3,ppm):172.5,166.5(c=o),163.6,132.1,131.7,130.2,129.4,128.6,128.3,128.1,128.0,127.9,127.0,122.1,113.7,63.5(ch2o),55.5,39.1(ch2nh),36.4,30.1,29.6,29.3,27.2,25.6,23.4,20.6,14.3(ch3).hrms(tof-ms,+):m/z[m+na]
+
calculated for c
32h43
nnao
4+
528.3084,found 528.3054。
[0084]
实施例9:化合物4-巯基苯甲酸-(二十二碳六烯酰胺乙基)-酯(j9)的制备
[0085]
化合物(j9)的结构式为:
[0086][0087]
将1.0mmol的二十二碳六烯酰乙醇胺和1.1mmol的4-巯基苯甲酰氯均匀混合溶于二氯甲烷中,在缚酸剂et3n的作用下,0-5℃下搅拌反应20h,反应结束后,1,2-二氯乙烷萃取两次,无水mgso4干燥,浓缩,快速硅胶柱层析纯化(v
乙酸乙酯
∶v
石油醚
=1∶1)和高效液相制备色谱进一步纯化,收集特征保留时间t
1/2
=10.99min的组分,浓缩即得淡黄色粘稠状产物j9。
[0088]
化合物(j9)的主要氢谱和碳谱数据如下:
[0089]
淡黄色粘稠状物,1h nmr(400mhz,cdcl3,ppm):7.95-7.93(m,2h,arh),7.30-7.26(m,2h,arh),5.92(s,1h,ch=ch),5.39-5.31(m,11h,ch=ch),4.43-4.40(m,2h,nhch2),3.68-3.64(m,2h,och2),2.86-2.83(m,8h,4ch2),2.53(s,3h,sch3),2.45-2.40(m,2h,ch2),2.28-2.25(m,2h,ch2),2.11-2.05(m,2h,ch2),1.86(br,1h,nh),1.31-1.27(m,2h,ch2),1.01-0.97(m,3h,ch3);
13
c nmr(100mhz,cdcl3,ppm):172.6,166.5(c=o),146.1,132.1,130.0,129.4,128.6,128.3,128.2,128.1,128.0,127.9,127.0,125.8,124.9,63.7(ch2o),
39.0(ch2nh),36.4,29.3,25.7,25.6,25.5,23.3,20.6,14.8,14.3(ch3).hrms(tof-ms,+):m/z[m+h]
+
calculated for c
32h44
no3s
+
522.3036,found 522.3019。
[0090]
实施例10:化合物4-乙氧基苯甲酸-(二十二碳六烯酰胺乙基)-酯(j10)的制备
[0091]
化合物(j10)的结构式为:
[0092][0093]
将1.0mmol的二十二碳六烯酰乙醇胺和1.1mmol的4-乙氧基苯甲酰氯均匀混合溶于四氢呋喃中,在缚酸剂pd的作用下,0-5℃下搅拌反应24h,反应结束后,1,2-二氯乙烷萃取两次,无水mgso4干燥,浓缩,快速硅胶柱层析纯化(v
乙酸乙酯
∶v
石油醚
=1∶1)和高效液相制备色谱进一步纯化,收集特征保留时间t
1/2
=10.64min的组分,浓缩即得淡黄色粘稠状产物j10。
[0094]
化合物(j10)的主要氢谱和碳谱数据如下:
[0095]
淡黄色粘稠状物,1h nmr(400mhz,cdcl3,ppm):8.00-7.98(m,2h,arh),6.94-6.91(m,2h,arh),5.98(s,1h,ch=ch),5.43-5.32(m,11h,ch=ch),4.41-4.39(m,2h,nhch2),4.13-4.08(m,2h,och2),3.68-3.64(m,2h,och2),2.87-2.83(m,8h,4ch2),2.43-2.40(m,2h,ch2),2.28-2.25(m,2h,ch2),2.11-2.05(m,2h,ch2),1.76(br,1h,nh),1.46-1.44(m,3h,ch3),1.31-1.29(m,2h,ch2),1.01-0.97(m,3h,ch3);
13
c nmr(100mhz,cdcl3,ppm):172.5,166.5(c=o),163.0,132.1,131.7,129.4,128.6,128.3,128.2,128.1,128.1,128.0,128.0,127.9,127.0,121.9,114.2,63.8(ch2o),63.5,39.1(ch2nh),36.4,30.2,29.5,25.7,25.6,25.5,23.4,20.6,14.7,14.3(ch3).hrms(tof-ms,+):m/z[m+na]
+
calculated for c
33h45
nnao
4+
542.3241,found 542.3228。
[0096]
实施例11:化合物3-苄基苯甲酸-(二十二碳六烯酰胺乙基)-酯(j11)的制备
[0097]
化合物(j11)的结构式为:
[0098][0099]
将1.0mmol的二十二碳六烯酰乙醇胺和1.1mmol的3-苄基苯甲酰氯均匀混合溶于乙腈中,在缚酸剂et3n的作用下,0-5℃下搅拌反应8h,反应结束后,1,2-二氯乙烷萃取两次,无水mgso4干燥,浓缩,快速硅胶柱层析纯化(v
乙酸乙酯
∶v
石油醚
=1∶1)和高效液相制备色谱进一步纯化,收集特征保留时间t
1/2
=13.60min的组分,浓缩即得淡黄色粘稠状产物j11。
[0100]
化合物(j11)的主要氢谱和碳谱数据如下:
[0101]
淡黄色粘稠状物,1h nmr(400mhz,cdcl3,ppm):7.67-7.65(m,2h,arh),7.48-7.46(m,2h,arh),7.44-7.40(m,2h,arh),7.38-7.36(m,2h,arh),7.23-7.22(m,1h,arh),5.87(s,1h,ch=ch),5.43-5.34(m,11h,ch=ch),4.44-4.41(m,2h,nhch2),3.69-3.67(m,2h,och2),2.88-2.84(m,10h,5ch2),2.44-2.41(m,2h,ch2),2.29-2.25(m,2h,ch2),2.11-2.08(m,2h,ch2),1.81-1.76(m,2h,ch2),1.32(br,1h,nh),1.01-0.97(m,3h,ch3);
13
c nmr(100mhz,cdcl3,ppm):172.6,166.5(c=o),158.8,136.5,132.1,131.1,129.6,129.4,128.7,128.6,128.5,128.3,128.2,128.2,128.1,128.1,128.0,128.0,127.9,127.0,127.6,127.0,122.3,120.3,115.4,70.2,64.0(ch2o),38.9(ch2nh),36.4,25.7,25.6,25.5,23.3,20.6,14.3(ch3).hrms(tof-ms,+):m/z[m+na]
+
calculated for c
38h47
nnao
3+
588.3448,found 588.3420。
[0102]
实施例12:化合物2-三氟甲基苯甲酸-(二十二碳六烯酰胺乙基)-酯(j12)的制备
[0103]
化合物(j12)的结构式为:
[0104][0105]
将1.0mmol的二十二碳六烯酰乙醇胺和1.1mmol的苯2-三氟甲基甲酰氯均匀混合溶于1,2-二氯乙烷中,在缚酸剂dea的作用下,0-5℃下搅拌反应12h,反应结束后,1,2-二氯乙烷萃取两次,无水mgso4干燥,浓缩,快速硅胶柱层析纯化(v
乙酸乙酯
∶v
石油醚
=1∶1)和高效液相制备色谱进一步纯化,收集特征保留时间t
1/2
=8.99min的组分,浓缩即得淡黄色粘稠状产物j12。
[0106]
化合物(j12)的主要氢谱和碳谱数据如下:
[0107]
淡黄色粘稠状物,1h nmr(400mhz,cdcl3,ppm):7.84-7.77(m,2h,arh),7.68-7.65(m,2h,arh),5.87(s,1h,ch=ch),5.44-5.31(m,11h,ch=ch),4.45-4.43(m,2h,nhch2),3.68-3.64(m,2h,och2),2.86-2.81(m,8h,4ch2),2.46-2.41(m,2h,ch2),2.28-2.25(m,2h,ch2),2.11-2.05(m,2h,ch2),1.83-1.57(m,2h,ch2),1.27(br,1h,nh),1.00-0.95(m,3h,ch3);
13
c nmr(100mhz,cdcl3,ppm):172.5,166.8(c=o),132.1,132.0,131.5,130.8,130.5,129.4,128.6,128.5,128.3,128.2,128.1,128.0,127.9,127.0,126.8,122.2,65.2(ch2o),38.4(ch2nh),36.3,25.7,25.6,25.5,23.3,20.6,14.3(ch3).hrms(tof-ms,+):m/z[m+na]
+
calculated for c
32h40
f3nnao
3+
566.2852,found 566.2842。
[0108]
实施例13:化合物3-腈基苯甲酸-(二十二碳六烯酰胺乙基)-酯(j13)的制备
[0109]
化合物(j13)的结构式为:
[0110][0111]
将1.0mmol的二十二碳六烯酰乙醇胺和1.1mmol的3-腈基苯甲酰氯均匀混合溶于四氢呋喃中,在缚酸剂pd的作用下,0-5℃下搅拌反应16h,反应结束后,1,2-二氯乙烷萃取两次,无水mgso4干燥,浓缩,快速硅胶柱层析纯化(v
乙酸乙酯
∶v
石油醚
=1∶1)和高效液相制备色谱进一步纯化,收集特征保留时间t
1/2
=8.04min的组分,浓缩即得淡黄色粘稠状产物j13。
[0112]
化合物(j13)的主要氢谱和碳谱数据如下:
[0113]
淡黄色粘稠状物,1h nmr(400mhz,cdcl3,ppm):8.34-8.28(m,2h,arh),7.88-7.86(m,1h,arh),7.64-7.60(m,1h,arh),5.89(s,1h,ch=ch),5.42-5.29(m,11h,ch=ch),4.48-4.45(m,2h,nhch2),3.71-3.67(m,2h,och2),2.86-2.80(m,8h,4ch2),2.45-2.41(m,2h,ch2),2.30-2.26(m,2h,ch2),2.12-2.05(m,2h,ch2),1.85-1.52(m,2h,ch2),1.27(br,1h,nh),1.00-0.97(m,3h,ch3);
13
c nmr(100mhz,cdcl3,ppm):172.7,164.7(c=o),136.2,139.8,133.3,132.1,131.1,129.6,129.5,128.6,128.4,128.3,128.1,128.0,127.9,127.8,127.0,117.8,113.1,64.6(ch2o),38.4(ch2nh),36.3,25.7,25.6,25.5,23.3,20.6,14.3(ch3).hrms(tof-ms,+):m/z[m+na]
+
calculated for c
32h40
n2nao
3+
523.2931,found 523.2928。
[0114]
实施例14:化合物2-甲基-5-氟苯甲酸-(二十二碳六烯酰胺乙基)-酯(j14)的制备
[0115]
化合物(j14)的结构式为:
[0116][0117]
将1.0mmol的二十二碳六烯酰乙醇胺和1.1mmol的2-甲基-5-氟苯甲酰氯均匀混合溶于二氧六环中,在缚酸剂et3n的作用下,0-5℃下搅拌反应20h,反应结束后,1,2-二氯乙烷萃取两次,无水mgso4干燥,浓缩,快速硅胶柱层析纯化(v
乙酸乙酯
∶v
石油醚
=1∶1)和高效液相制备色谱进一步纯化,收集特征保留时间t
1/2
=10.93min的组分,浓缩即得淡黄色粘稠状产物j14。
[0118]
化合物(j14)的主要氢谱和碳谱数据如下:
[0119]
淡黄色粘稠状物,1h nmr(400mhz,cdcl3,ppm):7.63-7.60(m,1h,arh),7.26-7.22(m,1h,arh),7.17-7.12(m,1h,arh),5.89(s,1h,ch=ch),5.39-5.31(m,11h,ch=ch),4.42-4.39(m,2h,nhch2),3.69-3.65(m,2h,och2),2.87-2.83(m,8h,4ch2),2.57(s,3h,ch3),2.46-2.41(m,2h,ch2),2.30-2.26(m,2h,ch2),2.11-2.05(m,2h,ch2),1.87-1.52(m,2h,ch2),1.30(br,1h,nh),1.01-0.97(m,3h,ch3);
13
c nmr(100mhz,cdcl3,ppm):172.6,166.3(c=o),161.8,159.3,136.2,136.1,133.3,132.1,130.3,129.5,128.3,128.2,128.1,128.0,127.9,119.4,117.3,63.9(ch2o),38.8(ch2nh),36.4,25.7,25.6,25.5,23.3,20.6,14.3(ch3).hrms(tof-ms,+):m/z[m+na]
+
calculated for c
32h42
fnnao
3+
530.3041,found 530.3032。
[0120]
实施例15:化合物2,4-二氯苯甲酸-(二十二碳六烯酰胺乙基)-酯(j15)制备
[0121]
化合物(j15)的结构式为:
[0122][0123]
将1.0mmol的二十二碳六烯酰乙醇胺和1.1mmol的2,4-二氯苯甲酰氯均匀混合溶于1,2-二氯乙烷中,在缚酸剂et3n的作用下,0-5℃下搅拌反应24h,反应结束后,1,2-二氯乙烷萃取两次,无水mgso4干燥,浓缩,快速硅胶柱层析纯化(v
乙酸乙酯
∶v
石油醚
=1∶1)和高效液相制备色谱进一步纯化,收集特征保留时间t
1/2
=12.85min的组分,浓缩即得淡黄色粘稠状产物j15。
[0124]
化合物(j15)的主要氢谱和碳谱数据如下:
[0125]
淡黄色粘稠状物,1h nmr(400mhz,cdcl3,ppm):7.84-7.82(m,1h,arh),7.51(s,1h,arh),7.36-7.33(m,1h,arh),5.86(s,1h,ch=ch),5.42-5.32(m,11h,ch=ch),4.45-4.43(m,2h,nhch2),3.70-3.66(m,2h,och2),2.87-2.83(m,10h,5ch2),2.45-2.40(m,2h,ch2),2.29-2.25(m,2h,ch2),2.13-2.05(m,2h,ch2),1.70(br,1h,nh),1.01-0.97(m,3h,ch3);
13
c nmr(100mhz,cdcl3,ppm):172.5,165.9(c=o),138.7,134.7,132.7,132.1,131.1,129.5,128.6,128.3,128.1,128.0,127.9,127.2,127.0,64.6(ch2o),38.6(ch2nh),36.4,25.7,25.6,25.5,23.3,20.6,14.3(ch3).hrms(tof-ms,+):m/z[m+na]
+
calculated for c
31h39
cl2nnao
3+
566.2199,found 566.2180。
[0126]
实施例16:化合物2,6-二氯苯甲酸-(二十二碳六烯酰胺乙基)-酯(j16)的制备
[0127]
化合物(j16)的结构式为:
[0128][0129]
将1.0mmol的二十二碳六烯酰乙醇胺和1.1mmol的2,6-二氯苯甲酰氯均匀混合溶于乙腈中,在缚酸剂et3n的作用下,0-5℃下搅拌反应8h,反应结束后,1,2-二氯乙烷萃取两次,无水mgso4干燥,浓缩,快速硅胶柱层析纯化(v
乙酸乙酯
∶v
石油醚
=1∶1)和高效液相制备色谱进一步纯化,收集特征保留时间t
1/2
=9.61min的组分,浓缩即得淡黄色粘稠状产物j16。
[0130]
化合物(j16)的主要氢谱和碳谱数据如下:
[0131]
淡黄色粘稠状物,1h nmr(400mhz,cdcl3,ppm):7.38-7.32(m,3h,arh),5.83(s,1h,ch=ch),5.45-5.32(m,11h,ch=ch),4.52-4.49(m,2h,nhch2),3.71-3.67(m,2h,och2),2.87-2.81(m,8h,4ch2),2.46-2.41(m,2h,ch2),2.28-2.24(m,2h,ch2),2.11-2.07(m,2h,ch2),1.82-1.57(m,2h,ch2),1.28(br,1h,nh),1.01-0.97(m,3h,ch3);
13
c nmr(100mhz,cdcl3,ppm):172.5,164.6(c=o),133.2,132.1,131.8,129.5,128.6,128.3,128.2,128.1,128.0,127.9,127.0,65.2(ch2o),38.5(ch2nh),36.4,25.7,25.6,25.5,23.3,20.6,14.3(ch3).hrms(tof-ms,+):m/z[m+na]
+
calculated for c
31h39
cl2nnao
3+
566.2199,found 566.2180。
[0132]
实施例17:化合物3-甲基-4-氟苯甲酸-(二十二碳六烯酰胺乙基)-酯(j17)的制备
[0133]
化合物(j17)的结构式为:
[0134][0135]
将1.0mmol的二十二碳六烯酰乙醇胺和1.1mmol的3-甲基-4-氟苯甲酰氯均匀混合溶于二氯甲烷中,在缚酸剂dea的作用下,0-5℃下搅拌反应16h,反应结束后,1,2-二氯乙烷萃取两次,无水mgso4干燥,浓缩,快速硅胶柱层析纯化(v
乙酸乙酯
∶v
石油醚
=1∶1)和高效液相制备色谱进一步纯化,收集特征保留时间t
1/2
=10.94min的组分,浓缩即得淡黄色粘稠状产物j17。
[0136]
化合物(j17)的主要氢谱和碳谱数据如下:
[0137]
淡黄色粘稠状物,1h nmr(400mhz,cdcl3,ppm):7.92-7.86(m,2h,arh),7.09-7.04(m,1h,arh),5.94(s,1h,ch=ch),5.41-5.31(m,11h,ch=ch),4.42-4.39(m,2h,nhch2),3.68-3.64(m,2h,och2),2.87-2.83(m,8h,4ch2),2.44-2.39(m,2h,ch2),2.33(s,3h,ch3),2.28-2.21(m,2h,ch2),2.19-2.11(m,2h,ch2),2.10-2.04(m,2h,ch2),1.30(br,1h,nh),1.00-0.96(m,3h,ch3);
13
c nmr(100mhz,cdcl3,ppm):172.6,166.0(c=o),163.3,133.4,132.0,129.6,129.5,129.4,128.6,128.3,128.1,128.0,127.9,127.0,125.6,125.4,125.2,115.3,63.8(ch2o),38.9(ch2nh),36.4,25.6,25.5,23.3,20.6,14.5(ch3),14.3(ch3).hrms(tof-ms,+):m/z[m+na]
+
calculated for c
32h42
fnnao
3+
530.3041,found 530.3032。
[0138]
实施例18:化合物正十六烷酸-(二十二碳六烯酰胺乙基)-酯(j18)的制备
[0139]
化合物(j18)的结构式为:
[0140]
[0141]
将1.0mmol的二十二碳六烯酰乙醇胺和1.1mmol的正十六烷酰氯均匀混合溶于四氢呋喃中,在缚酸剂et3n的作用下,0-5℃下搅拌反应24h,反应结束后,1,2-二氯乙烷萃取两次,无水mgso4干燥,浓缩,快速硅胶柱层析纯化(v
乙酸乙酯
∶v
石油醚
=1∶1)和高效液相制备色谱进一步纯化,收集特征保留时间t
1/2
=20.71min的组分,浓缩即得淡黄色粘稠状产物j18。
[0142]
化合物(j18)的主要氢谱和碳谱数据如下:
[0143]
淡黄色粘稠状物,1h nmr(400mhz,cdcl3,ppm):5.79(s,1h,ch=ch),5.42-5.35(m,11h,ch=ch),4.22-4.17(m,2h,nhch2),3.54-3.50(m,2h,och2),2.87-2.84(m,8h,4ch2),2.44-2.42(m,2h,ch2),2.37-2.33(m,2h,ch2),2.30-2.26(m,2h,ch2),2.11-2.09(m,2h,ch2),1.73(br,1h,nh),1.63-1.60(m,4h,2ch2),1.29-1.27(m,24h,12ch2),1.03-0.98(m,3h,ch3),0.91-0.89(m,3h,ch3);
13
c nmr(100mhz,cdcl3,ppm):174.0,172.4(c=o),132.1,129.4,128.6,128.3,128.1,127.9,127.0,63.1(ch2o),38.9(ch2nh),38.9,36.4,34.2,31.9,29.7,29.5,29.4,29.4,29.3,29.2,25.6,25.5,24.9,23.3,22.7,20.6,14.3,14.1(ch3).hrms(tof-ms,+):m/z[m+na]
+
calculated for c
40h67
nnao
3+
632.5013,found 632.4997。
[0144]
实施例19:取代苯甲酸二十二碳六烯酰胺乙基酯衍生物(j1-j18)的毒性评价
[0145]
将状态良好的细胞(raw264.7、h460和lo2中的一种)接种在96孔板中,3000-5000个/孔,培养过夜。待细胞贴壁后,分别加入工作浓度为10μm的化合物(j1-j18),作用时间为24h,并设置空白对照孔,每个化合物设置3个复孔。药物作用24h后,加入母液浓度为5mg/ml mtt,每孔10μl,mtt作用4h,吸走上清,不能吸走孔板底部的甲瓒,再向96孔板中加入dmso(120-180μl/孔)溶解甲瓒,利用分光光度仪在492nm处测量吸光度(od)值。利用测量得到的od值计算细胞存活率。实验结果见图4、图5、图6。
[0146]
实施例20:取代苯甲酸二十二碳六烯酰胺乙基酯衍生物(j1-j18)抑制raw264.7细胞中no的产生
[0147]
将巨噬细胞raw264.7接种于24孔板中,1.5万个/孔,培养过夜。待细胞贴壁后预先加入化合物给药处理2h,再每孔加入0.5μl的lps诱导24h,最终lps浓度为1μg/ml,地塞米松dex最终浓度为10μm,dhea的最终浓度为5μm,j1-j18化合物的最终浓度为5μm。收集细胞上清培养基作为检测样本。使用no检测试剂盒,在560nm处检测吸光度(od)值,检测raw264.7细胞分泌的no含量。实验结果表明,所检测的18个取代苯甲酸二十二碳六烯酰胺乙基酯衍生物都能抑制raw264.7细胞中no的产生。实验结果见表1、图7。
[0148]
实施例21:取代苯甲酸二十二碳六烯酰胺乙基酯衍生物(j1-j18)下调促炎介质tnf-α,il-6和il-1β的mrna表达
[0149]
将状态良好的raw264.7细胞接种在6孔板中,接种密度为4.5
×
105个每孔,细胞培养过夜,待细胞贴壁后加入化合物给药处理2h,j1-j18化合物作用终浓度为10μm,地塞米松dex作用终浓度为5μm,dhea的最终浓度为10μm,并加入lps诱导12h,lps的终浓度为1μg/ml。24h后,去除细胞培养基,提取rna,并对rna进行逆转录反应,得到的逆转录产物再进行pcr实验扩增,最后通过agilent ariamx pcr仪器的2-δδct
方法对实验结果进行定量。实验结果表明,所检测的18个取代苯甲酸二十二碳六烯酰胺乙基酯衍生物中j4,j9,j10,j11和j14化合物表现出显著下调促炎介质tnf-α,il-6和il-1β的mrna表达。实验结果见表1、图8、图9、图10。
[0150]
表1各组指标数据比较
[0151][0152][0153]
*p《0.05,**p《0.01,***p《0.001compare with only dheagroup.
[0154]
实施例22:取代苯甲酸二十二碳六烯酰胺乙基酯衍生物(j1-j18)抑制促炎性细胞因子tnf-α和il-1β的蛋白表达
[0155]
将状态良好的raw264.7细胞接种在6孔板中,接种密度为4.5
×
105个每孔,细胞培养过夜,待细胞贴壁后加入化合物给药预处理1h,j1-j18化合物作用终浓度为10μm,地塞米松dex作用终浓度为10μm,dhea的最终浓度为10μm,1h后再加入lps诱导24h,lps的终浓度为1μg/ml。24h后,去除细胞培养基,用蛋白裂解液裂解细胞,提取蛋白。把得到的蛋白样品用
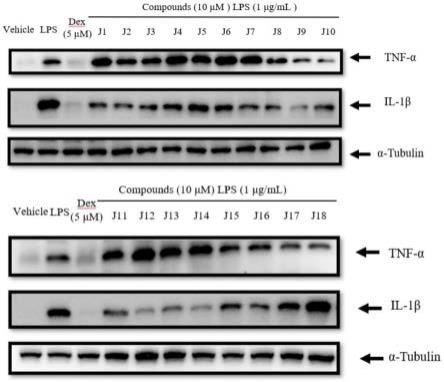
sds-page进行分析,在电转液的缓冲条件下,利用“三明治”法,将蛋白转移到pvdf膜上固定。电转后,用5%脱脂奶封闭2h,去除一些非特异性的蛋白干扰,再进行一抗,二抗孵育,二抗孵育结束后洗膜3次,每次10min。最后利用化学发光成像仪biorad进行曝光检测。实验结果表明,lps显著上调il-1β和tnf-α的蛋白表达,所检测的18个取代苯甲酸二十二碳六烯酰胺乙基酯衍生物中j2,j3,j8,j9,j10,j17和j18显著下调tnf-α的蛋白表达,而它们对il-1β的蛋白表达除了j18都显示出明显的下调趋势。实验结果见图11。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1