一种CSF-1R抑制剂中间体或其酸式盐的制备方法与流程
一种csf-1r抑制剂中间体或其酸式盐的制备方法
技术领域
1.本发明属于药物合成领域,具体涉及一种csf-1r抑制剂中间体或其酸式盐的制备方法。
背景技术:
2.csf-1r全称是细胞集落刺激因子-1受体,该受体为膜蛋白,表达于巨噬细胞和单核细胞的表面,其胞外段能够与巨噬细胞集落刺激因子结合,胞内段酪氨酸激酶可激活巨噬细胞及单核细胞下游细胞生长繁殖信号通路。因此,csf-1r信号通路对巨噬、单核细胞发育和分化,以及肿瘤相关巨噬细胞(tumor-associated macrophage,tam)的生理功能有重要影响。随着近年来肿瘤免疫治疗的进展,肿瘤相关巨噬细胞(tam)和骨髓源性抑制细胞(mdsc)被认为与肿瘤内部免疫抑制微环境的形成和支持肿瘤生长的血管生成有直接关系。同时,临床研究表明,tam的含量与肿瘤病人预后呈负相关。而在小鼠体内的药效实验证明,抑制csf-1r信号通路,能够显著降低肿瘤内部对免疫系统有抑制性的巨噬细胞数量,并提高cd8阳性的t细胞含量。这些实验结果表明,csf-1r小分子抑制剂可能会逆转肿瘤内部的免疫抑制环境,促进免疫系统的活化,并延长肿瘤患者的生命。
3.2018年,上海和誉生物医药有限公司在wo2018214867a1中公开了一系列具有选择性csf-1r抑制作用的化合物,在该申请中还公开了一系列中间体,其结构通式如下:
[0004][0005]
其中,该申请实施例1化合物用到的一个中间体,化学结构如下:其中文名称为:6-甲基-5-((2-(1-甲基-1h-吡唑-4-基)吡啶-4-基)氧基)吡啶-2-胺,该中间体的制备以5-溴-6-取代吡啶-2-胺原料,经四步反应得到,合成路线如下:
[0006][0007]
该合成方法步骤1)和2)的产率很低,分别只有45%和37.8%,四步反应总收率13.6%,且最后一步反应采用了铁粉还原法,该方法后处理过程中过滤困难,所生产的含芳
胺的铁泥和废水对环境污染严重,存在严重的技术缺陷,不适合工业化应用。因此,迫切需要研发出一种式(a)化合物的制备方法来进行工业化生产,以及制备出合格的api来满足临床研究以及上市药物制剂的生产需要。
技术实现要素:
[0008]
本发明的目的在于提供一种csf-1r抑制剂中间体或其酸式盐的制备方法,从而满足临床研究以及上市药物制剂的生产需要。
[0009]
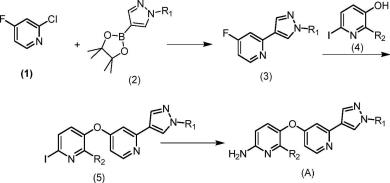
本发明第一方面提供一种式(a)化合物或其酸式盐的制备方法,包括如下合成步骤:
[0010]
步骤1)式(1)化合物或其酸式盐与式(2)化合物或其酸式盐在碱性条件下经缩合生成式(3)化合物或其酸式盐;
[0011]
步骤2)式(3)化合物或其酸式盐与式(4)化合物或其酸式盐在碱性条件下反应生成式(5)化合物或其酸式盐;
[0012]
步骤3)式(5)化合物或其酸式盐在碱性条件下氨化生成式(a)化合物或其酸式盐;反应路线如下:
[0013][0014]
各式中,r1选自氢、氘、c
1-4
烷基、c
3-6
环烷基和3-6元杂环基,上述基团任选进一步被一个或多个选自氘、羟基、卤素、氰基、c
1-4
烷基、c
3-6
环烷基、3-6元杂环基、c
1-4
烷氧基、羧基和氨基的取代基所取代;
[0015]
r2选自氢、氘、c
1-4
烷基和c
3-6
环烷基,上述基团任选进一步被一个或多个选自氘、羟基、卤素、氰基、c
1-4
烷基、c
3-6
环烷基、3-6元杂环基、c
1-4
烷氧基、羧基和氨基的取代基所取代;
[0016]
所述“杂环基”包含1-2个杂原子,选自氮原子或氧原子。
[0017]
作为优选的方案,所述的制备方法中r1选自氢、氘、甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、仲丁基、环丙基、环丁基、吗啉基、吡咯基、哌嗪基、氧杂环丁基和四氢呋喃基,上述基团任选进一步被一个或多个选自氘、羟基、卤素、氰基、甲基、乙基、正丙基、异丙基、环丙基、环丁基、吗啉基、吡咯基、氧杂环丁基、四氢呋喃基、甲氧基、乙氧基、异丙氧基、羧基和氨基的取代基所取代;
[0018]
r2选自氢、氘、甲基、三氟甲基、三氘甲基、乙基、异丙基、环丙基和环丁基。
[0019]
作为进一步优选的方案,所述的制备方法中r1选自氢、氘、甲基、三氟甲基、三氘甲基、甲氧甲基、羟甲基、乙基、甲氧乙基、羟乙基、正丙基、异丙基、正丁基、羟基取代异丁基、叔丁基、仲丁基、环丙基、环丁基、吗啉基、吗啉基乙基、吡咯基、甲基取代吡咯基、哌嗪基、氧杂环丁基和四氢呋喃基;
[0020]
r2选自氢、氘、甲基、乙基、三氟甲基和三氘甲基。
[0021]
作为进一步优选的方案,所述的制备方法中所述步骤2)中,式(3)化合物或其酸式盐和式(4)化合物或其酸式盐的投料摩尔比为1:(0.2~5)。
[0022]
作为更进一步优选的方案,所述的制备方法中所述步骤2)中,式(3)化合物或其酸式盐和式(4)化合物或其酸式盐的投料摩尔比为1:(0.5~3)。
[0023]
作为更进一步优选的方案,所述的制备方法中所述步骤2)中,式(3)化合物或其酸式盐和式(4)化合物或其酸式盐的投料摩尔比为1:(0.8~2)。
[0024]
作为进一步优选的方案,所述的制备方法中所述步骤2)在碱性条件下进行,所述碱性条件为无机碱体系,所述无机碱选自k2co3、khco3、cs2co3、na2co3或nahco3。
[0025]
作为进一步优选的方案,所述的制备方法中所述步骤3)中,式(5)化合物或其酸式盐与cf3conh2反应,投料摩尔比为1:(0.5~20)。
[0026]
作为更进一步优选的方案,所述的制备方法中所述步骤3)中,式(5)化合物或其酸式盐与cf3conh2反应,投料摩尔比为1:(1~10)。
[0027]
作为更进一步优选的方案,所述的制备方法中所述步骤3)中,式(5)化合物或其酸式盐与cf3conh2反应,投料摩尔比为1:(2~5)。
[0028]
作为进一步优选的方案,所述的制备方法中,所述步骤3)在碱性条件下进行,所述碱性条件为无机碱体系,所述无机碱选自k2co3、khco3、cs2co3、na2co3或nahco3。
[0029]
作为进一步优选的方案,所述的制备方法中所述步骤3)在催化剂催化下进行,所述催化剂为cui。
[0030]
作为进一步优选的方案,所述的制备方法中所述步骤1)在碱性条件下进行,所述碱性条件为无机碱体系,所述无机碱选自k2co3、khco3、cs2co3、na2co3或nahco3。
[0031]
作为进一步优选的方案,所述的制备方法中所述步骤1)采用pd(dppf)cl2作为缩合剂。
[0032]
作为优选的方案,所述的制备方法中所述酸式盐包括无机酸盐或有机酸盐。
[0033]
作为进一步优选的方案,所述无机酸盐选自盐酸盐、硫酸盐、氢溴酸盐、氢氟酸盐、氢碘酸盐或磷酸盐。
[0034]
作为进一步优选的方案,所述有机酸盐选自醋酸盐、二氯醋酸盐、三氯醋酸盐、三氟乙酸盐、苯磺酸盐、对甲苯磺酸盐、4-氯苯磺酸盐、1,5-萘二磺酸盐、萘-2-磺酸盐、乙烷-1,2-二磺酸盐、甲磺酸盐、乙磺酸盐、苯甲酸盐、癸酸盐、己酸盐、辛酸盐、肉桂酸盐、柠檬酸盐、环己烷氨基磺酸盐、樟脑磺酸盐、天门冬氨酸盐、樟脑酸盐、葡萄糖酸盐、葡糖醛酸盐、谷氨酸盐、异抗坏血酸盐、乳酸盐、苹果酸盐、扁桃酸盐、焦谷氨酸盐、酒石酸盐、十二烷基硫酸盐、二苯甲酰酒石酸盐、蚁酸盐、富马酸盐、半乳糖酸盐、龙胆酸盐、乙酰氧肟酸盐、丙二酸盐、丁二酸盐、戊二酸盐、己二酸盐、癸二酸盐、2-酮戊二酸盐、乙醇酸盐、马尿酸盐、羟乙基磺酸盐、乳糖酸盐、抗坏血酸盐、天冬氨酸盐、月桂酸盐、马来酸盐、烟酸盐、油酸盐、乳清酸盐、草酸盐、棕榈酸盐、双羟萘酸盐、丙酸盐、4-乙酰氨基苯甲酸盐、4-氨基苯甲酸盐、水杨酸
盐、4-氨基水杨酸盐、2,5-二羟基苯甲酸盐、1-羟基-2-萘甲酸盐、硬脂酸盐、硫氰酸盐、十一碳烯酸盐或琥珀酸盐。
[0035]
本发明第二方面提供一种式(5)化合物:
[0036][0037]
其中,r1选自氢、氘、c
1-4
烷基、c
3-6
环烷基和3-6元杂环基,上述基团任选进一步被一个或多个选自氘、羟基、卤素、氰基、c
1-4
烷基、c
3-6
环烷基、3-6元杂环基、c
1-4
烷氧基、羧基和氨基的取代基所取代;
[0038]
r2选自氢、氘、c
1-4
烷基和c
3-6
环烷基,上述基团任选进一步被一个或多个选自氘、羟基、卤素、氰基、c
1-4
烷基、c
3-6
环烷基、3-6元杂环基、c
1-4
烷氧基、羧基和氨基的取代基所取代;
[0039]
所述“杂环基”包含1-2个杂原子,选自氮原子或氧原子。
[0040]
作为优选的方案,所述式(5)化合物中r1选自氢、氘、甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、仲丁基、环丙基、环丁基、吗啉基、吡咯基、哌嗪基、氧杂环丁基和四氢呋喃基,上述基团任选进一步被一个或多个选自氘、羟基、卤素、氰基、甲基、乙基、正丙基、异丙基、环丙基、环丁基、吗啉基、吡咯基、氧杂环丁基、四氢呋喃基、甲氧基、乙氧基、异丙氧基、羧基和氨基的取代基所取代;
[0041]
r2选自氢、氘、甲基、三氟甲基、三氘甲基、乙基、异丙基、环丙基和环丁基。
[0042]
作为优选的方案,所述式(5)化合物中r1选自氢、氘、甲基、三氟甲基、三氘甲基、甲氧甲基、羟甲基、乙基、甲氧乙基、羟乙基、正丙基、异丙基、正丁基、羟基取代异丁基、叔丁基、仲丁基、环丙基、环丁基、吗啉基、吗啉基乙基、吡咯基、甲基取代吡咯基、哌嗪基、氧杂环丁基和四氢呋喃基;
[0043]
r2选自氢、氘、甲基、乙基、三氟甲基和三氘甲基。
[0044]
作为优选的方案,所述式(5)化合物具有式(5’)化合物结构:
[0045][0046]
本发明第三方面提供式(1’)化合物、式(2’)化合物或式(3’)化合物:
[0047][0048]
其中,r1选自氢、氘、c
1-4
烷基、c
3-6
环烷基和3-6元杂环基,上述基团任选进一步被一个或多个选自氘、羟基、卤素、氰基、c
1-4
烷基、c
3-6
环烷基、3-6元杂环基、c
1-4
烷氧基、羧基
和氨基的取代基所取代;
[0049]
r2选自氢、氘、c
1-4
烷基和c
3-6
环烷基,上述基团任选进一步被一个或多个选自氘、羟基、卤素、氰基、c
1-4
烷基、c
3-6
环烷基、3-6元杂环基、c
1-4
烷氧基、羧基和氨基的取代基所取代;
[0050]
所述“杂环基”包含1-2个杂原子,选自氮原子或氧原子。
[0051]
作为优选的方案,所述的式(1’)化合物、式(2’)化合物或式(3’)化合物中r1选自氢、氘、甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、仲丁基、环丙基、环丁基、吗啉基、吡咯基、哌嗪基、氧杂环丁基和四氢呋喃基,上述基团任选进一步被一个或多个选自氘、羟基、卤素、氰基、甲基、乙基、正丙基、异丙基、环丙基、环丁基、吗啉基、吡咯基、氧杂环丁基、四氢呋喃基、甲氧基、乙氧基、异丙氧基、羧基和氨基的取代基所取代;
[0052]
r2选自氢、氘、甲基、三氟甲基、三氘甲基、乙基、异丙基、环丙基和环丁基。
[0053]
作为优选的方案,所述式(1’)化合物、式(2’)化合物或式(3’)化合物中r1选自氢、氘、甲基、三氟甲基、三氘甲基、甲氧甲基、羟甲基、乙基、甲氧乙基、羟乙基、正丙基、异丙基、正丁基、羟基取代异丁基、叔丁基、仲丁基、环丙基、环丁基、吗啉基、吗啉基乙基、吡咯基、甲基取代吡咯基、哌嗪基、氧杂环丁基和四氢呋喃基;
[0054]
r2选自氢、氘、甲基、乙基、三氟甲基和三氘甲基。
[0055]
与现有技术相比,本发明具有以下几方面的技术优势:
[0056]
1、在环保层面,本发明在步骤2)中利用碘取代中间体氨化,避免了采用铁粉还原法,极大减小了环保压力。
[0057]
2、在技术层面,本发明采用三步反应得到式(a)化合物,各步反应产率均较高,三步总收率达到60%,解决了现有技术需四步反应且收率不足15%的药物可及性问题,且本发明各步产物纯度很高,稳定性好,有利于工业化应用。
[0058]
3、在操作层面,本发明采用原料或试剂简单易得,反应条件温和,可操作性强,整个制备过程中没有用到柱层析技术,适合工业化生产。
[0059]
4、本发明工艺路线还提供了一种新的碘取代中间体,该中间体稳定性高,可用于制备式(a)化合物或其酸式盐。
[0060]
5、本发明工艺路线还提供了一种式(a)化合物酸式盐,优选为三氟乙酸盐、盐酸盐或氢碘酸盐。
附图说明
[0061]
图1为具体实施例第三步合成产物的hplc图谱。
具体实施方式
[0062]
本技术的发明人经过广泛而深入地研究,开发出一种csf-1r抑制剂中间体或其酸式盐的制备方法。所述制备方法以2-氯-4-氟吡啶为原料,经三步反应制备得到。本发明制备方法操作简单,各步收率很高,且工艺稳定,可适应工业化生产需求,解决药物可及性问题,有利于加速csf-1r抑制剂的临床开发和药物上市。在此基础上,完成了本发明。
[0063]
下面结合实施例对本发明做进一步详细、完整地说明,但决非限制本发明,本发明也并非仅局限于实施例的内容。
[0064]
本发明的化合物结构是通过核磁共振(nmr)来确定的。nmr化学位移(δ)以百万分之一(ppm)的单位给出。nmr的测定是用bruker biospin gmbh 600核磁共振仪,测定溶剂为氘代二甲基亚砜(dmso-d6),氘代甲醇(cd3od)和氘代氯仿(cdcl3),内标为四甲基硅烷(tms)。
[0065]
薄层层析硅胶板使用烟台黄海hsgf254或青岛gf254硅胶板,tlc采用的规格是0.15mm~0.20mm。
[0066]
本发明的化合物纯度是通过安捷伦高效液相色谱(hplc)来检测的。色谱柱:waters c18。流动相a:0.05%三氟乙酸水溶液,流动相b:0.05%三氟乙酸乙腈溶液。流速:1.0ml/min。检测波长:254nm。
[0067]
本发明实施例中的起始原料是已知的并且可以在市场上买到,或者可以采用或按照本领域已知的方法来合成。
[0068]
在无特殊说明的情况下,本发明的所有反应均在干燥氮气或氩气氛下进行,溶剂为干燥溶剂,反应温度单位为摄氏度(℃)。
[0069]
具体实施例
[0070]
第一步:4-氟-2-(1-甲基-1h-吡唑-4-基)吡啶的合成
[0071][0072]
氮气保护下加入异丙醇(24l,316.2mol),2-氯-4-氟吡啶(4.0kg,30.4mol),1-甲基-4-(4,4,5,5-四甲基-1,3,2-二恶英-2-基)-1h吡唑(6.64kg,31.9mol),k2co3(12.17kg,88.1mol),水(1.2kg,66.7mol)和pd(dppf)cl2(111.3g,0.15mol),将反应混合物加热至80-90℃,搅拌反应12小时,tlc检测原料反应完毕,将反应液冷却至50℃,加入乙酰半胱氨酸(400ml),继续搅拌1小时,再将反应液冷却至20℃,加入水(10l),分出有机相,浓缩至干。在残余物中加入甲苯(10l)和水(10l),加入盐酸调ph值至1,分液,用乙酸乙酯萃取水相,在水相中加入na2co3调节ph值至9,然后再用乙酸乙酯萃取(12l*3),合并有机相,浓缩至干,加入甲基叔丁基醚(10l),过滤,滤饼干燥得到4-氟-2-(1-甲基-1h-吡唑-4-基)吡啶4.3kg(收率:79.63%,纯度:99.74%)。
[0073]
第二步:6-碘-2-甲基-3-((2-(1-甲基-1h-吡唑-4-基)吡啶-4-基)氧基)吡啶的合成
[0074][0075]
氮气保护下加入n-甲基吡咯烷酮(150ml,1.56mol),4-氟-2-(1-甲基-1h-吡唑-4-基)吡啶(39.58g,0.22mol),6-碘-2-甲基吡啶-3-醇(50.0g,0.21mol)和na2co3(45.1g,0.43mol),将反应混合物加热至100-110℃,搅拌反应60小时,tlc检测原料反应完毕,将反应液冷却至室温,加入乙醇(300ml),继续搅拌10分钟。控温35℃以下逐滴加反应液至500ml水中,继续搅拌2小时,过滤,滤饼干燥得6-碘-2-甲基-3-((2-(1-甲基-1h-吡唑-4-基)吡
啶-4-基)氧基)吡啶71.73g(收率:82%,纯度:99.46%)。
[0076]
esi[m+h]
+
393.2。
[0077]1hnmr(dmso-d6)δ8.39(d,j=8.0,1h),8.28(s,1h),7.99(s,1h),7.75(d,j=8.0,1h),7.32(d,j=8.0,1h),7.24(d,j=8.0,1h),6.69(dd,j=5.6,2.4,1h),3.86(s,3h),2.34(s,3h)。
[0078]
第三步:6-甲基-5-((2-(1-甲基-1h-吡唑-4-基)吡啶-4-基)氧基)吡啶-2-胺的合成
[0079][0080]
氮气保护下加入1,4-二氧六环(300ml,3.39mol),二甲基甲酰胺(35ml,0.42mol),6-碘-2-甲基-3-((2-(1-甲基-1h-吡唑-4-基)吡啶-4-基)氧基)吡啶(50g,0.13mol),cf3conh2(50.44g,0.45mol),k2co3(61.6g,0.45mol),cui(1.21g,0.006mol)和二甲基乙二胺(5.62g,0.064mol),将反应混合物加热至100-110℃,搅拌反应1小时,将反应液冷却至70℃,加入纯水150ml,继续搅拌1小时,将反应混合液减压浓缩有至100-150ml,将反应液冷却至室温,加入20-30%(w/w)nh
3.
h2o(30ml),水(300ml),继续搅拌1小时,用二氯甲烷萃取(800ml*2),分液,水相用二氯甲烷(400ml)萃取,合并有机相,用饱和食盐水洗涤3次(100ml*3),控温40℃下减压浓缩至100-150ml体积,加入甲基叔丁基醚(250ml),打浆搅拌2小时,过滤,滤饼干燥得6-甲基-5-((2-(1-甲基-1h-吡唑-4-基)吡啶-4-基)氧基)吡啶-2-胺33.2g(收率92.2%,纯度:94.53%,hplc谱图见附图1)。
[0081]
ms m/z(esi):282[m+h]
+
。
[0082]
从附图1可知该步反应目标产物6-甲基-5-((2-(1-甲基-1h-吡唑-4-基)吡啶-4-基)氧基)吡啶-2-胺粗品纯度达94.53%,产物粗品中含有少量杂质,经图谱解析,主要杂质化学结构如下:
[0083][0084]
在本发明提及的所有文献都在本技术中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本发明所涉及的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1