一种特戈拉赞中间体及其制备方法与应用与流程
1.本发明涉及原料药合成技术领域,尤其涉及一种特戈拉赞中间体及其制备方法与应用。
背景技术:
2.特戈拉赞,又名替戈拉生、tegoprazan、cj-12420,于2018年7月获韩国食品药品安全部(mfds)批准上市,用于胃食管反流疾病和糜烂性食管炎的治疗。tegoprazan是一种竞争性钾离子酸阻滞剂(p-cab)和氢离子/钾离子交换atp酶(h+/k+atpase)抑制剂,起效快,可长时间控制胃液ph值,是一款全新的用于治疗胃食管反流病及糜烂性食管炎的药物。
3.特戈拉赞(tegoprazan)化学名为(s)-4-((5,7-二氟色满-4-基)氧)-n,n,2-三甲基-1h-苯并咪唑-6-甲酰胺,化学结构中含有苯并咪唑结构和手性5,7-二氟色满-4-氧基结构。特戈拉赞的制备主要涉及到(s)-5,7-二氟-3,4-二氢-2h-色原烯-4-醇的制备和4-羟基-n,n,2-三甲基-1h-苯并咪唑-6-甲酰胺的制备及其缩合反应。专利cn101341149b公开了tegoprazan的制备方法,具体为4-羟-n,n,2-三甲基-1-[(4-甲苯基)磺酰基]-1h-苯并[d]咪唑-6-甲酰胺和(s)-5,7-二氟-3,4-二氢-2h-色原烯-4-醇在三丁基膦/addp作用下发生缩合反应,制备得到(-)4-[((4s)-5,7-二氟-3,4-2h-色原烯-4-基)氧基]-n,n,2-三甲基-1-[(4-甲苯基)磺基]-1h-苯并[d]咪唑-6-甲酰胺中间体,后者在碱作用下脱除保护基完成tegoprazan的制备,基于上述专利的描述,tegoprazan的制备主要涉及到4-羟基-n,n,2-三甲基-1-[(4-甲苯基)磺酰基]-1h-苯并[d]咪唑-6-甲酰胺和(s)-5,7-二氟-3,4-二氢-2h-色原烯-4-醇的缩合反应,该缩合反应不仅涉及到使用危险的试剂三丁基膦和偶氮类化合物,且收率较低,成本较高。
[0004]
因此,开发新的适合产业化、具有成本优势的合成tegoprazan的中间体,可以降低tegoprazan产业化生产的危险性,成为本领域亟需。
技术实现要素:
[0005]
有鉴于此,本发明提供了一种特戈拉赞中间体及其制备方法与应用,解决了目前常用中间体制备特戈拉赞需要添加危险试剂,工艺成本高,且产率低的问题。
[0006]
为了达到上述目的,本发明采用如下技术方案:
[0007]
本发明提供一种特戈拉赞中间体的制备方法,包括如下步骤:
[0008]
(1)将2-氨基-3-氯苯酚与苄胺混合后进行取代反应,得到2-氨基-3-苯胺酚;
[0009]
(2)将2-氨基-3-苯胺酚与乙酸酐混合后进行酰胺化反应,得到n-[2-羟基-6-(苄胺基)苯基]乙酰胺;
[0010]
(3)将n-[2-羟基-6-(苄胺基)苯基]乙酰胺、还原剂、酸性溶剂混合后进行环合反应,得到1-苄基-4-羟基-2-甲基-1h-苯并咪唑;
[0011]
(4)将1-苄基-4-羟基-2-甲基-1h-苯并咪唑、双(三氯甲基)碳酸酯、路易斯酸、溶剂混合后进行反应,得到1-苄基-4-羟基-2-甲基-1h-苯并咪唑-6-羧酸;
[0012]
(5)将1-苄基-4-羟基-2-甲基-1h-苯并咪唑-6-羧酸、n,n-二甲基甲酰胺、氯化亚砜、二甲胺溶液混合后进行催化反应,得到1-苄基-4-羟基-n,n,2-三甲基-1h-苯并[d]咪唑-6-羧酰胺;
[0013]
所述1-苄基-4-羟基-n,n,2-三甲基-1h-苯并[d]咪唑-6-羧酰胺为特戈拉赞中间体。
[0014]
作为优选,所述步骤(1)中,取代反应在碱性溶剂中进行;所用碱性溶剂为碳酸钠、碳酸氢钠、氢氧化钠、乙醇钠或甲醇钠;2-氨基-3-氯苯酚与碱性溶剂的质量比为1:0.6~0.8。
[0015]
作为优选,所述步骤(1)中,2-氨基-3-氯苯酚与苄胺的质量比为1:0.5~1.2;取代反应的温度为40~60℃,取代反应的时间为2~4h。
[0016]
作为优选,所述步骤(2)中,乙酸酐与2-氨基-3-氯苯酚的质量比为1:1.2~2;酰胺化反应的温度为60~100℃,酰胺化反应的时间为1~2h。
[0017]
作为优选,所述步骤(3)中,还原剂为原甲酸三乙酯,酸性溶剂为盐酸;还原剂与2-氨基-3-氯苯酚的质量比为2~3:1,酸性溶剂与2-氨基-3-氯苯酚的摩尔比为1.5~3:1;环合反应的温度为0~10℃,环合反应的时间为1~10h。
[0018]
作为优选,所述步骤(4)中,路易斯酸为三氯化铝、氯化锌或溴化铁,溶剂为二氯甲烷、三氯甲烷或二氯乙烷;路易斯酸与2-氨基-3-氯苯酚的质量比为1.1~1.3:1,双(三氯甲基)碳酸酯与2-氨基-3-氯苯酚的质量比为3~4.5:1,2-氨基-3-氯苯酚与溶剂的质量体积比为1g:4~6ml。
[0019]
作为优选,所述步骤(4)中,反应的温度为-30~-1℃,反应的时间为1.5~4h。
[0020]
作为优选,所述步骤(5)中,氯化亚砜与2-氨基-3-氯苯酚的质量比为1~1.1:1,n,n-二甲基甲酰胺用量是2-氨基-3-氯苯酚用量的2~3%,n,n-二甲基甲酰胺与二甲胺溶液的质量比为1:80~100,二甲胺溶液为质量浓度为30~50%的二甲胺水溶液;催化反应的温度为20~40℃,催化反应的时间为10~15h。
[0021]
本发明还提供了所述特戈拉赞中间体的制备方法制备得到的特戈拉赞中间体,所述特戈拉赞中间体为1-苄基-4-羟基-n,n,2-三甲基-1h-苯并[d]咪唑-6-羧酰胺;所述1-苄基-4-羟基-n,n,2-三甲基-1h-苯并[d]咪唑-6-羧酰胺的结构式为
[0022]
本发明还提供了所述特戈拉赞中间体在制备特戈拉赞中的应用。
[0023]
经由上述的技术方案可知,与现有技术相比,本发明有益效果如下:
[0024]
(1)本发明所述反应操作简单,使用的试剂安全性高,适合产业化合成特戈拉赞及关键中间体;
[0025]
(2)本发明采用多步反应制备得到特戈拉赞中间体1-苄基-4-羟基-n,n,2-三甲基-1h-苯并[d]咪唑-6-羧酰胺,反应选择性高,收率高。
附图说明
[0026]
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据提供的附图获得其他的附图。
[0027]
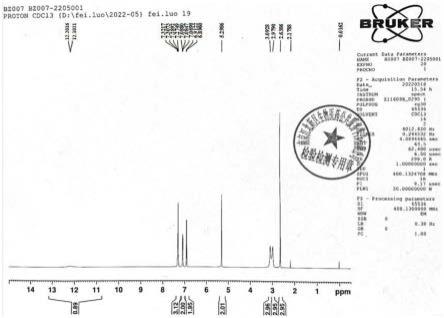
图1为本发明实施例1所得1-苄基-4-羟基-n,n,2-三甲基-1h-苯并咪唑-6-羧酰胺的氢谱图。
具体实施方式
[0028]
本发明提供一种特戈拉赞中间体的制备方法,包括如下步骤:
[0029]
(1)将2-氨基-3-氯苯酚与苄胺混合后进行取代反应,得到2-氨基-3-苯胺酚;
[0030]
(2)将2-氨基-3-苯胺酚与乙酸酐混合后进行酰胺化反应,得到n-[2-羟基-6-(苄胺基)苯基]乙酰胺;
[0031]
(3)将n-[2-羟基-6-(苄胺基)苯基]乙酰胺、还原剂、酸性溶剂混合后进行环合反应,得到1-苄基-4-羟基-2-甲基-1h-苯并咪唑;
[0032]
(4)将1-苄基-4-羟基-2-甲基-1h-苯并咪唑、双(三氯甲基)碳酸酯、路易斯酸、溶剂混合后进行反应,得到1-苄基-4-羟基-2-甲基-1h-苯并咪唑-6-羧酸;
[0033]
(5)将1-苄基-4-羟基-2-甲基-1h-苯并咪唑-6-羧酸、n,n-二甲基甲酰胺、氯化亚砜、二甲胺溶液混合后进行催化反应,得到1-苄基-4-羟基-n,n,2-三甲基-1h-苯并[d]咪唑-6-羧酰胺;
[0034]
所述1-苄基-4-羟基-n,n,2-三甲基-1h-苯并[d]咪唑-6-羧酰胺为特戈拉赞中间体。
[0035]
在本发明中,所述步骤(1)中,取代反应在碱性溶液中进行;所用碱性溶剂优选为碳酸钠、碳酸氢钠、氢氧化钠、乙醇钠或甲醇钠,进一步优选为碳酸钠或碳酸氢钠;2-氨基-3-氯苯酚与碱性试剂的质量比优选为1:0.6~0.8,进一步优选为1:0.65~0.75。
[0036]
在本发明中,所述步骤(1)中,2-氨基-3-氯苯酚与苄胺混合前,将2-氨基-3-氯苯酚与溶剂混合,所述溶剂为乙酸乙酯。
[0037]
在本发明中,所述步骤(1)中,2-氨基-3-氯苯酚与苄胺的质量比优选为1:0.5~1.2,进一步优选为1:0.8~1;取代反应的温度优选为40~60℃,进一步优选为50~55℃;取代反应的时间优选为2~4h,进一步优选为2.5~3h。
[0038]
在本发明中,所述步骤(2)中,乙酸酐与2-氨基-3-氯苯酚的质量比优选为1:1.2~2,进一步优选为1:1.1~1.5;酰胺化反应的温度优选为60~100℃,进一步优选为80~95℃;酰胺化反应的时间优选为1~2h,进一步优选为1.5h。
[0039]
在本发明中,所述步骤(3)中,还原剂为原甲酸三乙酯,酸性溶剂为盐酸;还原剂与2-氨基-3-氯苯酚的质量比为2~3:1,进一步优选为2.5:1;酸性溶剂与2-氨基-3-氯苯酚的摩尔比优选为1.5~3:1,进一步优选为2.5:1;环合反应的温度优选为0~10℃,进一步优选为6~8℃;环合反应的时间优选为1~10h,进一步优选为8~9h。
[0040]
在本发明中,所述步骤(4)中,路易斯酸优选为三氯化铝、氯化锌或溴化铁,进一步优选为氯化锌或溴化铁;溶剂优选为二氯甲烷、三氯甲烷或二氯乙烷,进一步优选为三氯甲
烷或二氯乙烷;路易斯酸与2-氨基-3-氯苯酚的质量比为1.1~1.3:1,进一步优选为1.2:1;双(三氯甲基)碳酸酯与2-氨基-3-氯苯酚的质量比为3~4.5:1,进一步优选为3.5~4:1;2-氨基-3-氯苯酚与溶剂的质量体积比优选为1g:4~6ml,进一步优选为1g:4.5~5.5ml。
[0041]
在本发明中,所述步骤(4)中,反应的温度优选为-30~-1℃,进一步优选为-25~-5℃;反应的时间优选为1.5~4h,进一步优选为2~3h。
[0042]
在本发明中,所述步骤(5)中,氯化亚砜与2-氨基-3-氯苯酚的质量比为1~1.1:1,进一步优选为1.05:1;n,n-二甲基甲酰胺用量优选为2-氨基-3-氯苯酚用量的2~3%,进一步优选为2-氨基-3-氯苯酚用量的2.5%;n,n-二甲基甲酰胺与二甲胺溶液的质量比优选为1:80~100,进一步优选为1:90~95;二甲胺溶液为质量浓度为30~50%的二甲胺水溶液;催化反应的温度优选为20~40℃,进一步优选为25~30℃;催化反应的时间优选为10~15h,进一步优选为12~14h。。
[0043]
在本发明中,所述步骤(1)的反应式为:
[0044][0045]
在本发明中,所述步骤(2)的反应式为:
[0046][0047]
在本发明中,所述步骤(3)的反应式为:
[0048][0049]
在本发明中,所述步骤(4)的反应式为:
[0050][0051]
在本发明中,所述步骤(5)的反应式为:
[0052][0053]
本发明还提供了所述特戈拉赞中间体的制备方法制备得到的特戈拉赞中间体,所述特戈拉赞中间体为1-苄基-4-羟基-n,n,2-三甲基-1h-苯并[d]咪唑-6-羧酰胺;所述1-苄基-4-羟基-n,n,2-三甲基-1h-苯并[d]咪唑-6-羧酰胺的结构式为
[0054]
本发明还提供了所述特戈拉赞中间体在制备特戈拉赞中的应用。
[0055]
下面结合实施例对本发明提供的技术方案进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
[0056]
实施例1
[0057]
(1)将5g2-氨基-3-氯苯酚溶于25ml乙酸乙酯中,加入3.43g碳酸氢钠搅拌,并滴加5.18g苄胺,经过0.5h滴加结束,之后反应2h,反应结束后水洗有机相,最后将有机相浓缩得到2-氨基-3苯胺酚;
[0058]
(2)将上步所得2-氨基-3苯胺酚与9.48g乙酸酐在80℃下反应1h得n-[2-羟基-6-(苄胺基)苯基]乙酰胺;
[0059]
(3)在n-[2-羟基-6-(苄胺基)苯基]乙酰胺中加入13.77g原甲酸三乙酯,在0℃下滴加2.5g质量浓度为32%的盐酸,反应1h得到1-苄基-4-羟基-2-甲基-1h-苯并咪唑;
[0060]
(4)将6.44g三氯化铝加入至25ml三氯甲烷中,之后加入22.05g双(三氯甲基)碳酸酯后,在-20℃下加入1-苄基-4-羟基-2-甲基-1h-苯并咪唑,在-30~-1℃下反应2h后得到1-苄基-4-羟基-2-甲基-1h-苯并咪唑-6-羧酸;
[0061]
(5)将1-苄基-4-羟基-2-甲基-1h-苯并咪唑-6-羧酸在0.13gn,n-二甲基甲酰胺催化下,加入5.30g氯化亚砜和13.5g质量浓度为30%的二甲胺溶液,在30℃下反应12h得到1-苄基-4-羟基-n,n,2-三甲基-1h-苯并咪唑-6-羧酰胺。
[0062]
实施例2
[0063]
(1)将5g2-氨基-3-氯苯酚溶于25ml乙酸乙酯中,加入3.88g碳酸钠搅拌,并滴加4.10g苄胺,经过0.5h滴加结束,之后反应2h,反应结束后水洗有机相,最后将有机相浓缩得到2-氨基-3苯胺酚;
[0064]
(2)将上步所得2-氨基-3苯胺酚与7.11g乙酸酐在80℃下反应1h(时间)得n-[2-羟基-6-(苄胺基)苯基]乙酰胺;
[0065]
(3)在n-[2-羟基-6-(苄胺基)苯基]乙酰胺中加入10.32g原甲酸三乙酯,在0℃下滴加2.5g质量浓度为32%的盐酸,反应1h得到1-苄基-4-羟基-2-甲基-1h-苯并咪唑;
[0066]
(4)将5.57g三氯化铝加入至25ml二氯乙烷中,之后加入16.54g双(三氯甲基)碳酸酯后,在-15℃下加入1-苄基-4-羟基-2-甲基-1h-苯并咪唑,在-30~-1℃下反应2h后得到
1-苄基-4-羟基-2-甲基-1h-苯并咪唑-6-羧酸;
[0067]
(5)将1-苄基-4-羟基-2-甲基-1h-苯并咪唑-6-羧酸在0.15gn,n-二甲基甲酰胺催化下,加入5.30g氯化亚砜和13.5g质量浓度为40%的二甲胺,在30℃下反应12h得到1-苄基-4-羟基-n,n,2-三甲基-1h-苯并咪唑-6-羧酰胺。
[0068]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1