一种力致荧光增强聚合物及其制备方法
1.本发明属于化学技术领域,具体地说,涉及一种力致荧光增强聚合物及其制备方法。
背景技术:
2.压(力)致变色是指荧光颜色或/和强度等在外加机械力(摩擦、剪切等)或静压力作用下发生可逆变化的现象。静压力致变色(piezochromicfluorescence,pcf)材料因为其荧光在压力作用下能连续改变,在压力传感系统(如深海潜水器和飞行器风洞试验等)、光学记录、防伪、信息显示和存储等领域拥有很大的应用潜力。
3.目前报道的静压力致变色材料主要有:日本科学家yamaguchi教授制备出“螺旋桨”型噻吩衍生物,晶体发出微弱黄色荧光。在3.2gpa静压力作用下其荧光完全淬灭,相对于初始态光谱产生53nm的红移(j.am. chem.soc.2013,135,1032)。吉林大学田文晶教授课题组设计、合成了高发光的蒽烯衍生物,在从1atm到7.9gpa压力刺激下荧光光谱连续红移,红移增大至124nm(angew.chem.int.ed.2012,51,10782)。虽然目前有大量的力致变色材料被报道,但是依然存在以下问题:(1)力致变色材料都是粉末,不具有力学强度,很难被实际应用;(2)pcf材料在压力作用下荧光是逐渐变暗的,降低了材料的识别度。
技术实现要素:
4.有鉴于此,本发明提供了一种力致荧光增强聚合物及其制备方法。
5.为了解决上述技术问题,本发明公开了一种力致荧光增强聚合物的制备方法,包括以下步骤:
6.步骤1:硼配位化合物e(i)的合成:
7.其合成路线如下:
[0008][0009]
称取3,4,5-三甲氧基苯甲醛(ii)和三(五氟苯)硼烷(iii,b(c6f5)3)倒入研钵中,使用研杵在一定的温度下研磨一定的时间。薄层板示踪反应进程,待反应完全后,通过柱层色谱提纯,获得白色粉末,即为最终产物硼配位化合物(i);
[0010]
步骤2:将硼配位化合物(i)粉末溶解于有机溶剂中,加入聚合物颗粒,进行超声,同时不断加入相应的有机溶剂直至溶解,最终获得硼配位化合物和有机溶剂混合溶液;将其旋涂,干燥成膜,得到硼配位化合物的聚甲基丙烯酸甲酯薄膜,即为力致荧光增强聚合
物;
[0011]
进一步地,3,4,5-三甲氧基苯甲醛和b(c6f5)3的摩尔比为1∶1.0-1∶1.1;
[0012]
进一步地,反应时间为0.1-1h。
[0013]
进一步地,反应温度为30-70℃。
[0014]
进一步地,硼配位化合物与溶剂的质量体积比(mg/ml)为1∶10-50∶10,硼配位化合物与聚合物颗粒的质量比(g/g)为0.1∶1000-5∶100;
[0015]
进一步地,聚合物为聚甲基丙烯酸甲酯(pmma)和氢化苯乙烯-丁二烯嵌段共聚物(sebs),对应的有机溶剂为氯仿和四氢呋喃(thf)。
[0016]
本发明还公开了一种上述的制备方法制备得到的力致荧光增强聚合物。
[0017]
与现有技术相比,本发明可以获得包括以下技术效果:
[0018]
本发明的力致荧光增强聚合物其具有力学性质好(该聚合物制备的薄膜可弯曲、拉伸和剪切等)、成本低和高发光效率的特点,关键是该材料通过荧光的强度能定量识别外界压力,可用于压力传感系统。其次,硼配化合物的合成采用固相研磨法,不需要溶剂,产率高,对环境无污染。
[0019]
当然,实施本发明的任一产品并不一定需要同时达到以上所述的所有技术效果。
附图说明
[0020]
此处所说明的附图用来提供对本发明的进一步理解,构成本发明的一部分,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。在附图中:
[0021]
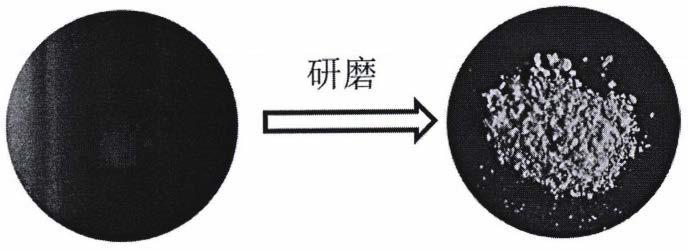
图1是本发明3,4,5-三甲氧基苯甲醛(ii)与三(五氟苯)硼烷(iii)混合物荧光照片和研磨制备的硼配位化合物的荧光照片;
[0022]
图2是本发明掺杂量为1%的pmma薄膜(1%tobs@pmma)在不同压力下的荧光光谱;tobs为硼配位化合物的缩写。
[0023]
图3是本发明掺杂量为5%的sebs薄膜(5%tobs@pmma)在不同压力下的荧光光谱;tobs为硼配位化合物的缩写。
具体实施方式
[0024]
以下将配合实施例来详细说明本发明的实施方式,藉此对本发明如何应用技术手段来解决技术问题并达成技术功效的实现过程能充分理解并据以实施。
[0025]
本发明公开了一种力致比率变色聚合物的制备方法,包括以下步骤:
[0026]
步骤1,硼配位化合物(i)的合成:
[0027]
其合成路线如下:
[0028][0029]
称取3,4,5-三甲氧基苯甲醛(ii)和三(五氟苯)硼烷(iii)倒入研钵中,使用研杵
充分研磨。其中,3,4,5-三甲氧基苯甲醛和b(c6f5)3的摩尔比为 1∶1-1∶1.1,研磨反应时间为0.5-1h。研磨反应温度为30-70℃。薄层板示踪反应进程,待反应完全后,通过柱层色谱提纯,获得白色粉末,即为最终产物硼配位化合物(i)。其中,(ii)分子量196g/mol;(i)分子量512.0g/mol; (iii)分子量708.2g/mol;
[0030]
步骤2:将硼配位化合物(i)固体粉末溶解于氯仿溶剂中,然后加入聚甲基丙烯酸甲酯(pmma)颗粒,其中硼配位化合物固体粉末与氯仿溶剂的质量体积比(mg/ml)为1∶10-50∶10,硼配位化合物固体粉末与聚甲基丙烯酸甲酯颗粒的质量比(g/g)为0.1∶1000-5∶100;进行超声,同时不断加入氯仿溶剂直至溶解,最终获得硼配位化合物和聚甲基丙烯酸甲酯(pmma)混合溶液,其中硼配位化合物固体粉末的聚甲基丙烯酸甲酯颗粒的掺杂量为 0.1
‰‑
5%。将其旋涂,干燥成膜,得到硼配位化合物的聚甲基丙烯酸甲酯薄膜,即为力致荧光增强聚合物,该薄膜都能拉伸、弯曲和剪切,具有较强的荧光。
[0031]
大部分荧光分子很贵,而聚合物高分子很便宜,本发明最大掺杂5%,这样可以大大降低成本。
[0032]
实施例1
[0033]
一种力致比率变色聚合物的制备方法,包括以下步骤:
[0034]
步骤1:硼配位化合物(i)的合成:
[0035]
称取3,4,5-三甲氧基苯甲醛(ii)0.20g(10mmol)和三(五氟苯)硼烷(iii) 0.51g(10mmol)倒入研钵中。在45℃环境温度下,使用研杵研磨0.5小时。然后将粗产物溶解到乙酸乙酯中,随后加入无水硫酸镁进行干燥。过滤、减压浓缩得到的剩余物进行硅胶柱层析分离,洗脱剂为(石油醚/乙酸乙酯=10∶1),通过减压蒸去有机溶剂获得白色粉末硼配位化合物(i)0.64g,总产率为90.4%。
[0036]
硼配位化合物(i)的核磁表征数据为:1h nmr(500mhz,cdcl3)δ9.19 (s,1h),δ7.72(s,1h),3.83(s,9h)。
[0037]
13
c nmr(125mhz,cdcl3);δ191.0,153.6,148.6,145.0,143.3,137.3, 132.6,111.3,106.0,60.8,56.1。
[0038]
步骤2:将硼配位化合物(i)0.01g溶解于10ml的氯仿(cdcl3)溶剂中,加入聚甲基丙烯酸甲酯(pmma)颗粒1g,进行超声,同时不断加入氯仿溶液直至溶解,最终获得掺杂量为1%的硼配位化合物pmma混合溶液,将其旋涂,干燥成膜,得到硼配位化合物的聚甲基丙烯酸甲酯薄膜。即为力致荧光增强聚合物,该薄膜都能拉伸、弯曲和剪切,具有较强的发光效率。
[0039]
实施例2
[0040]
步骤1:称取3,4,5-三甲氧基苯甲醛(ii)0.20g(10mmol)和三(五氟苯) 硼烷(iii)0.56g(11mmol)倒入研钵中。在30℃环境温度下,使用研杵充分研磨0.5h。然后将粗产物溶解到乙酸乙酯中,随后加入无水硫酸镁进行干燥。过滤、减压浓缩得到的剩余物进行硅胶柱层析分离,洗脱剂为(石油醚/乙酸乙酯=10∶1),通过减压蒸去有机溶剂获得白色粉末硼配位化合物(i) 0.59g,总产率为83.3%。
[0041]
步骤2:将硼配位化合物(i)0.05g溶解于10ml的氯仿(cdcl3)溶剂中,加入聚甲基丙烯酸甲酯(pmma)颗粒10g,进行超声,同时不断加入氯仿溶液直至溶解,最终获得掺杂量为0.5%的硼配位化合物pmma混合溶液,将其旋涂,干燥成膜,得到硼配位化合物的聚甲基
丙烯酸甲酯薄膜,即为力致荧光增强聚合物。该薄膜都能拉伸、弯曲和剪切,具有较强的发光效率。如图2所示,随着压力的增加,荧光的强度不增加,0.9gpa时达到最大值。
[0042]
实施例3
[0043]
步骤1:称取3,4,5-三甲氧基苯甲醛(ii)0.60g(30mmol)和三(五氟苯) 硼烷(iii)1.53g(30mmol)倒入研钵中。在70℃环境温度下,使用研杵充分研磨1h。然后将粗产物溶解到乙酸乙酯中,随后加入无水硫酸镁进行干燥。过滤、减压浓缩得到的剩余物进行硅胶柱层析分离,洗脱剂为(石油醚/ 乙酸乙酯=10∶1),通过减压蒸去有机溶剂获得白色粉末硼配位化合物(i) 2.0g,总产率为94.3%。
[0044]
步骤2:将硼配位化合物(i)0.05g溶解于10ml的氯仿(cdcl3)溶剂中,加入聚甲基丙烯酸甲酯(pmma)颗粒10g,进行超声,同时不断加入氯仿溶液直至溶解,最终获得掺杂量为5
‰
的硼配位化合物pmma混合溶液,将其旋涂,干燥成膜,得到硼配位化合物的聚甲基丙烯酸甲酯薄膜,即为力致荧光增强聚合物。该薄膜都能拉伸、弯曲和剪切,具有较强的发光效率。
[0045]
实施例4
[0046]
步骤1:称取3,4,5-三甲氧基苯甲醛(ii)0.20g(10mmol)和三(五氟苯) 硼烷(iii)0.51g(10mmol)倒入研钵中。在70℃环境温度下,使用研杵充分研磨0.1h。然后将粗产物溶解到乙酸乙酯中,随后加入无水硫酸镁进行干燥。过滤、减压浓缩得到的剩余物进行硅胶柱层析分离,洗脱剂为(石油醚/乙酸乙酯=10∶1),通过减压蒸去有机溶剂获得白色粉末硼配位化合物 (i)0.45g,总产率为63.4%。
[0047]
步骤2:将硼配位化合物粉末(i)0.2g溶解于10ml的thf溶剂中,加入氢化苯乙烯-丁二烯嵌段共聚物(sebs)颗粒100g,进行超声,同时不断加入thf溶液直至溶解,最终获得掺杂量为0.2%的二苯乙烯腈衍生物 sebs混合溶液,将其旋涂,干燥成膜,该薄膜都能拉伸、弯曲和剪切,具有微弱荧光。
[0048]
实施例5
[0049]
步骤1:称取3,4,5-三甲氧基苯甲醛(ii)1.96g(0.1mol)和三(五氟苯) 硼烷(iii)5.12g(0.1mol)倒入研钵中。在70℃环境温度下,使用研杵充分研磨0.6h。然后将粗产物溶解到乙酸乙酯中,随后加入无水硫酸镁进行干燥。过滤、减压浓缩得到的剩余物进行硅胶柱层析分离,洗脱剂为(石油醚/乙酸乙酯=10∶1),通过减压蒸去有机溶剂获得白色粉末硼配位化合物(i) 6.86g,总产率为96.9%。
[0050]
步骤2:将硼配位化合物(i)0.05g溶解于10ml的thf溶剂中,加入氢化苯乙烯-丁二烯嵌段共聚物(sebs)颗粒1g,进行超声,同时不断加入 thf溶液直至溶解,最终获得硼配位化合物掺杂量为5%的sebs混合溶液,将其旋涂,干燥成膜,该薄膜都能拉伸、弯曲和剪切,具有较强的荧光。然后截取合适尺寸放入压腔,进行压力传感测试。如图3所示,随着压力增加荧光变强。
[0051]
实施例6
[0052]
步骤1:步骤1、称取3,4,5-三甲氧基苯甲醛(ii)0.20g(10mmol)和三(五氟苯)硼烷(iii)0.56g(11mmol)倒入研钵中。在50℃环境温度下,使用研杵充分研磨0.3h。然后将粗产物溶解到乙酸乙酯中,随后加入无水硫酸镁进行干燥。过滤、减压浓缩得到的剩余物进行硅胶柱层析分离,洗脱剂为(石油醚/乙酸乙酯=10∶1),通过减压蒸去有机溶剂获得白色粉末
硼配位化合物(i)0.49g,总产率为69.0%。
[0053]
步骤2:将硼配位化合物粉末(i)0.02g溶解于10ml的thf溶剂中,加入氢化苯乙烯-丁二烯嵌段共聚物(sebs)颗粒1g,进行超声,同时不断加入thf溶液直至溶解,最终获得掺杂量为2%的二苯乙烯腈衍生物sebs 混合溶液,将其旋涂,干燥成膜,该薄膜都能拉伸、弯曲和剪切,具有较强的荧光。
[0054]
上述说明示出并描述了发明的若干优选实施例,但如前所述,应当理解发明并非局限于本文所披露的形式,不应看作是对其他实施例的排除,而可用于各种其他组合、修改和环境,并能够在本文所述发明构想范围内,通过上述教导或相关领域的技术或知识进行改动。而本领域人员所进行的改动和变化不脱离发明的精神和范围,则都应在发明所附权利要求的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1