一种N-N-双-恶唑烷酮生物碱类化合物、制备方法及在医药领域的应用与流程
一种n-n-双-恶唑烷酮生物碱类化合物、制备方法及在医药领域的应用
技术领域
1.本发明属于恶唑烷酮生物碱类化合物技术领域,具体涉及一种n-n-双-恶唑烷酮生物碱类化合物、所述化合物的制备方法、包含所述生物碱类化合物的药物组合物及其在医药领域的应用。
背景技术:
2.公开该背景技术部分的信息仅仅旨在增加对本发明的总体背景的理解,而不必然被视为承认或以任何形式暗示该信息构成已经成为本领域一般技术人员所公知的现有技术。
3.生物碱类化合物是一类含氮的碱性有机化合物,是天然产物中重要的一类有效成分,广泛分布于毛茛科、罂粟科、防己科、茄科、夹竹桃科、芸香科、豆科、小檗科等植物中,根据生物碱基本结构的不同可分为60多类,如有机胺类、吡咯烷类、吡啶类等。文献调研发现生物碱的药理活性众多,主要包括抗肿瘤、抗炎、镇痛、抗菌、抗病毒等,该类化合物有良好的开发应用前景。
4.忍冬(lonicera japonica thunb.)为忍冬科(caprifoliaceae)忍冬属(lonicera)多年生半常绿缠绕灌木,是一种具有悠久历史的常用中药,其花、茎、叶均可入。忍冬叶为忍冬科植物忍冬的干燥叶,即金银花叶,质地较厚,叶对生,呈卵形、椭圆形或长椭圆形,被覆绒毛。作为金银花的副产品,产量较大。现代研究表明忍冬叶中含有环烯醚萜类、黄酮类、有机酸、挥发油等化学成分,药理活性实验发现忍冬叶具有抗病原微生物、抗炎、抗氧化、降血脂等作用。文献报道金银花中存在生物碱类成分,而忍冬叶中生物碱类成分未见报道。
技术实现要素:
5.为了充分发掘忍冬叶的药效物质基础,本发明对忍冬叶化学成分进行了研究,从中得到新型结构的n-n-双-恶唑烷酮生物碱类同分异构体,且所得化合物具有显著的抗氧化和保肝活性,有望应用于化药的开发。
6.基于上述技术成果,本发明提供以下技术方案:
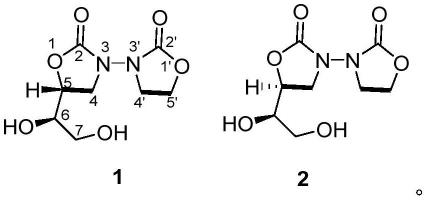
7.本发明第一方面,提供一种n-n-双-恶唑烷酮生物碱类化合物,所述化合物具有如下式1或式2所示的结构:
8.9.上述第一方面所述n-n-双-恶唑烷酮生物碱类化合物,还包括式1或式2所示化合物药学上可接受的盐、酯、溶剂化物、代谢产物或前药;其中,上述化合物药学上可接受的或酯主要包括为了改善所述化合物物理、化学性质所进行的成盐或成酯修饰,例如,为了改善水溶性等。
10.本发明第二方面,提供第一方面所述n-n-双-恶唑烷酮生物碱类化合物的制备方法,以忍冬叶乙醇提取物为原料分离得到,所述分离包括如下步骤:
11.(1)采用大孔树脂对忍冬叶乙醇提取物进行梯度洗脱,洗脱液依次为水、35~45%乙醇、55~65%乙醇、75~85%乙醇和92~98%乙醇,获取水洗脱部分,加入硅胶拌样;
12.(2)制备硅胶柱色谱,所述柱色谱中硅胶采用二氯甲烷混匀后装柱;将上述步骤(1)中拌样后的硅胶加入柱色谱,并采用二氯甲烷-甲醇进行梯度洗脱,所述洗脱液中二氯甲烷、甲醇两试剂的洗脱梯度依次为13~17:1、8~12:1、4~6:1、0.8~1.2:1(v/v);合并上述各部分洗脱液并浓缩依次得到fr.1~fr.4四个部分;
13.(3)将上述fr.1部分加入甲醇溶液溶解,通过mci中压制备色谱进行分离,以甲醇-水体系进行梯度洗脱,上述洗脱液中甲醇的浓度依次为4~6%、13~17%、23~27%、33~37%、96~100%;收集上述洗脱液通过hplc进行检查将相似馏分进行合并,得到fr.1-1~fr.1-10共十个组分,取组分fr.1-1通过半制备色谱进行分离纯化,流动相为乙腈-0.0.08~0.12%甲酸水,得到化合物1和化合物2.
14.上述步骤(1)中,一些优选的实施方式如下:
15.所述忍冬叶乙醇提取物的制备方法如下:向粉碎后的忍冬叶中加入30~50%乙醇溶液进行加热回流,固液比为1:2.5~3.5,将回流提取的醇溶液合并过滤并浓缩至无醇味,得到上述忍冬叶提取物。
16.上述制备方法中,回流的加热温度为95~105℃;回流次数及回流提取时间可根据药材提取情况常规确定,考虑到提取效率,所述回流提取的次数可以为三次,每次回流时间为1.8~2.2h。
17.所述大孔树脂采用弱极性吸附型大孔树脂,进一步的,为苯乙烯型弱极性共聚体,具体的实例如ab-8大孔树脂。
18.另外,上述步骤(1)中,所述水洗脱部分与硅胶以质量比1:0.8~1.2进行拌样。
19.上述步骤(2)中,所述洗脱液中二氯甲烷及甲醇的体积比为15:1、10:1、5:1、1:1,或为16:1、11:1、6:1、1.2:1,或14:1、9:1、4:1、0.8:1.
20.上述步骤(3)中,一些优选的技术方案如下:
21.所述洗脱液中,甲醇的溶度依次为5%、15%、25%、35%、100%,或为4%、13%、23%、33%、96%,或为6%、16%、26%、36%、100%。
22.所述流动相中,乙腈与甲酸水溶液的体积比为8~12:88~92,进一步的,为10:90.
23.本发明第三方面,提供一种药物组合物,所述组合物中,包括活性剂量的第一方面所述n-n-双-恶唑烷酮生物碱类化合物。
24.上述第三方面所述药物组合物可行的施用方式包括但不限于用于药物制剂、营养品或护肤品的制备,上述药物制剂、营养品或护肤品的其中一个功效在于预防、改善或治疗过氧化途径相关的疾病,另一个功效在于预防、改善、修复或治疗肝损伤及肝损伤相关疾病。其中,过氧化途径相关疾病包括但不限于肿瘤、糖尿病及其并发症、血管硬化、心脑血管
疾病、肾病、辐射损伤或免疫性疾病;所述肝损伤或肝损伤相关疾病包括但不限于药物原因、酒精原因或其他疾病因素导致的肝损伤。
25.优选的方案中,所述药物组合物中还包括药学上可接受的载体;所述药物载体包括但不限于赋形剂、润滑剂、粘合剂、崩解剂、溶剂、增溶剂、悬浮剂、等渗剂、缓冲剂、舒缓剂、防腐剂、抗氧化剂、着色剂、甜味剂或其他添加剂等。
26.本发明第四方面,提供第一方面所述n-n-双-恶唑烷酮生物碱类化合物、第三方面所述药物组合物在医药领域的应用。
27.上述第四方面所述医药领域的应用,其应用方式包括至少包括以下几种:
28.(1)将上述n-n-双-恶唑烷酮生物碱类化合物、药物组合物应用于制备药物制剂、营养品、食品或护肤品;
29.(2)将活性剂量的上述n-n-双-恶唑烷酮生物碱类化合物、药物组合物施用于有需求的个体以实现预防、改善疾病症状或治疗疾病的作用,所述疾病为过氧化相关疾病或肝部疾病;所述施用手段为剂量可精确调控的给药方式,包括手术、口服、注射或介入等;
30.(3)将上述n-n-双-恶唑烷酮生物碱类化合物、药物组合物应用于疾病模型的制备,如过氧化抑制模型或肝损伤抑制模型。
31.以上一个或多个技术方案的有益效果是:
32.提供了上述从忍冬叶中分离纯化的两个新n-n-双-恶唑烷酮生物碱同分异构体的结构鉴定方法及其制备方法,以忍冬叶为原料,来源广泛,制备工艺简单,经济、安全,得率高,所得的两个新n-n-双-恶唑烷酮生物碱同分异构体具有较好的抗氧化和保肝活性,而且毒副作用低,具有良好的药用前景。
附图说明
33.构成本发明的一部分的说明书附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。
34.图1为化合物1的1h nmr图谱;
35.图2为化合物1的
13
c nmr图谱;
36.图3为化合物1的hmqc图谱;
37.图4为化合物1的hmbc图谱;
38.图5为化合物1的noesy图谱;
39.图6为化合物1的icd谱;
40.图7为化合物1的hresims图谱;
41.图8为化合物2的1h nmr图谱;
42.图9为化合物2的
13
c nmr图谱;
43.图10为化合物2的hmqc图谱;
44.图11为化合物2的hmbc图谱;
45.图12为化合物2的noesy图谱;
46.图13为化合物2的icd谱;
47.图14为化合物2的hresims图谱。
具体实施方式
48.应该指出,以下详细说明都是例示性的,旨在对本发明提供进一步的说明。除非另有指明,本文使用的所有技术和科学术语具有与本发明所属技术领域的普通技术人员通常理解的相同含义。
49.需要注意的是,这里所使用的术语仅是为了描述具体实施方式,而非意图限制根据本发明的示例性实施方式。如在这里所使用的,除非上下文另外明确指出,否则单数形式也意图包括复数形式,此外,还应当理解的是,当在本说明书中使用术语“包含”和/或“包括”时,其指明存在特征、步骤、操作、器件、组件和/或它们的组合。
50.为了使得本领域技术人员能够更加清楚地了解本发明的技术方案,以下将结合具体的实施例详细说明本发明的技术方案。
51.实施例1
52.式1或式2所示结构的化合物,其制备方法具体步骤如下:
53.(1)取忍冬叶药材10kg,粉碎,40%乙醇加热回流提取三次,固液比为1:3,每次2h,将提取液合并过滤,浓缩至无醇味,得忍冬叶乙醇提取物1.2kg。
54.(2)采用ab-8大孔树脂柱色谱对忍冬叶乙醇提取物进行分离,依次采用水、40%乙醇、60%乙醇、80%乙醇和95%乙醇进行梯度洗脱,每个梯度洗脱体积为30l,合并各部分洗脱液,分别浓缩后得水、40%、60%、80%和95%乙醇洗脱部分。
55.(3)取水部位洗脱样品(1kg)与硅胶1:1拌匀备用,采用硅胶柱色谱进行分离,5kg硅胶填料用15l二氯甲烷混匀后装柱,密封静置4h,用1.5l二氯甲烷将水部位硅胶拌样样品匀后的上样,静置1h,依次采用二氯甲烷-甲醇(15:1、10:1、5:1、1:1,v/v)进行洗脱,每个梯度洗脱体积为20l,合并各部分洗脱液,分别浓缩后得4个部位(fr.1~4)。
56.(4)取fr.1(80g)用甲醇水溶解成50ml溶液,采用mci中压制备色谱(柱体积1500ml)进行分离,以甲醇-水体系(5%、15%、25%、35%、100%)进行梯度洗脱,每1500ml洗脱液为一馏分,每个梯度洗脱体积为4500ml,用hplc对各馏分进行检测,将成分峰组成类似的馏分进行合并,共得到10段(fr.1-1~fr.1-10),取fr.1-1(2.5g)采用岛津半制备色谱进行分离纯化,流动相为乙腈-0.1%甲酸水(10:90,v/v,288nm),得到化合物1(20mg)和化合物2(32mg)。
57.结构鉴定:对分离得到的单体成分应用bruker impact ii质谱仪和burker 400mhz核磁共振波谱仪分别进行hr-esi-ms,nmr谱的测定,所得核磁数据见表1,鉴定该两个新n-n-双-恶唑烷酮生物碱同分异构体的结构。
58.式1所示化合物的结构认定
59.(5r,6r)-5-(1,2-二羟基乙基)-[3,3'-双恶唑烷]-2,2'-二酮((5r,6r)-5-(1,2-dihydroxyethyl)-[3,3'-bioxazolidine]-2,2'-dione)(1):黄色油状物;hr-esi-ms:m/z 231.0558[m-h]-(理论值:231.0612,c8h
11
n2o6),确定其分子式为:c8h
12
n2o6,不饱和度为4。红外光谱在3361cm-1
处有吸收表明存在羟基官能团,2932cm-1
处有吸收表明可能存在-ch2基团,1737cm-1
处的吸收表明存在羰基官能团。
[0060]1h-nmr谱显示4组连氧氢信号δ
h 4.28(1h,m,h-5)、δ
h 4.26(1h,m,h-6)、δh3.55(2h,dd,j=3.6,6.4hz,h-7)、δh3.61(2h,t,j=6.4hz,h-5'),以及2组连n氢信号δ
h 2.81(1h,dd,j=17.6,6.4hz,h-4a)、δ
h 2.23(1h,dd,j=17.6,2.0hz,h-4b)、δ
h 2.35(2h,t,j=
6.4hz,h-4')。
13
c-nmr谱显示存在8个碳信号,包括2个羰基碳信号δc176.7(c-2)、δc173.6(c-2'),2个连氧ch信号δ
c 88.7(c-5)和δc68.2(c-6),4个ch2信号(包括2个连氧ch2信号δc61.2(c-7)和δc57.5(c-5'),2个连n ch2信号δc38.4(c-4)和δc38.2(c-4'))。通过1h-nmr和
13
c-nmr谱推断该化合物含有oxazolidin-2-one和乙二醇的结构片段,并通过二维谱图进一步确定该推断。在hmbc谱中,δh(h-4)与δ
c 173.6(c-2)、δ
c 88.7(c-5)、δ
c 68.2(c-6)相关,δ
h 4.28(h-5)与δ
c 176.7(c-2)、δ
c 38.4(c-4)、δ
c 68.2(c-6)、δ
c 61.2(c-7)相关,δ
h 4.26(h-6)与δ
c 88.7(c-5)、δ
c 38.4(c-4)、δ
c 61.2(c-7)相关,δ
h 3.55(h
2-7)与δ
c 88.7(c-5)、δ
c 68.2(c-6)相关,δ
h 2.35(h-4')与δ
c 173.6(c-2')、δ
c 88.7(c-5')相关,δ
h 3.61(h-5')与δ
c 173.6(c-2')、δ
c 38.2(c-4')相关,进一步确定化合物21含有1个oxazolidin-2-one结构和1个5-(1,2-dihydroxyethyl)oxazolidin-2-one结构。根据化合物2的分子式确定oxazolidin-2-one和5-(1,2-dihydroxyethyl)oxazolidin-2-one结构片段通过n-n相连,最终确定化合物1的平面结构为5-(1,2-dihydroxyethyl)-[3,3'-bioxazolidine]-2,2'-dione。
[0061]
化合物1的立体结构通过与mo2(oac)4试剂反应后测定圆二色谱确定。通过反应40min后的cd谱可观察到320nm处为正手征性,表明二醇体系的二面角扭角为顺时针,进而确定c-6位构型为r。进一步通过noe谱中相关信号:h-5与h-4b相关,h-6与h-4a相关,确定c-5位构型为r。综上所述,化合物1的结构确定为(5r,6r)-5-(1,2-二羟基乙基)-[3,3'-双恶唑烷]-2,2'-二酮((5r,6r)-5-(1,2-dihydroxyethyl)-[3,3'-bioxazolidine]-2,2'-dione)。
[0062]
式2所示化合物的结构认定
[0063]
(5s,6r)-5-(1,2-二羟基乙基)-[3,3'-双恶唑烷]-2,2'-二酮((5s,6r)-5-(1,2-dihydroxyethyl)-[3,3'-bioxazolidine]-2,2'-dione)(2):黄色油状物;hr-esi-ms:m/z 231.0557[m-h]-(理论值:231.0612,c8h
11
n2o6),确定其分子式为:c8h
12
n2o6,不饱和度为4。红外光谱在3341cm-1
处有吸收表明存在羟基官能团,2938cm-1
处有吸收表明可能存在-ch2基团,1753cm-1
处的吸收表明存在羰基官能团。
[0064]1h-nmr谱显示4组连氧氢信号δ
h 4.29(1h,m,h-5)、δ
h 4.29(1h,m,h-6)、δh3.53(2h,dd,j=3.6,6.4hz,h-7)、δ
h 3.62(2h,t,j=6.4hz h-5'),以及2组连n氢信号δ
h 2.80(1h,dd,j=17.6,6.4hz,h-4a)、2.23(1h,dd,j=17.6,2.0hz,h-4b)、δ
h 2.35(1h,t,j=6.4hz,h-4')。
13
c-nmr谱显示存在8个碳信号,包括2个羰基碳信号δc176.7(c-2)、δc173.6(c-2'),2个连氧ch信号δc88.7(c-5)和δc68.2(c-6),4个ch2信号(包括2个连氧ch2信号δc61.2(c-7)和δc57.5(c-5'),2个连n ch2信号δc38.4(c-4)和δc38.2(c-4'))。通过比较化合物1和2的核磁谱图表明两者为同分异构体。化合物2的立体结构通过与mo2(oac)4试剂反应后测定圆二色谱确定。通过反应40min后的cd谱可观察到320nm处为正手征性,表明二醇体系的二面角扭角为顺时针,进而确定c-6位相对构型为r。进一步通过noe谱中相关信号:h-5与h-4a相关,h-6与h-4b相关,确定c-5位构型为s。综上所述,化合物2的结构确定为(5s,6r)-5-(1,2-二羟基乙基)-[3,3'-双恶唑烷]-2,2'-二酮((5s,6r)-5-(1,2-dihydroxyethyl)-[3,3'-bioxazolidine]-2,2'-dione)。
[0065]
表1化合物1和2的1h nmr(400mhz,dmso-d6)和
13
c nmr数据(100mhz,dmso-d6)
[0066][0067][0068]
药效活性验证
[0069]
一、式1、式2所示化合物的抗氧化和保肝活性研究
[0070]
1、偶氮二异丁脒盐酸盐(aaph)诱导肝细胞氧化应激损伤模型的建立及抗氧化活性筛选
[0071]
hepg2细胞株采用rpmi-1640培养基、10%胎牛血清(fbs)进行培养,置于5%co2,37℃的细胞培养箱中培养至细胞覆盖率达90%以上时用于传代。取96孔细胞培养板,每孔加入浓度为3
×
105个/ml的细胞悬液100μl,在37℃、5%co2培养箱中贴壁培养24h后,吸出原培养液弃去。设置空白组、aaph模型组、阳性对照组、给药组,进行研究。空白组加100μl无血清的培养基,aaph模型组给予100μl 100mmol/l浓度的aaph(无血清的培养基溶解),阳性对照组给予100μl 50μmol/l槲皮素,给药组给予100μl 50μmol/l化合物1和2,每个样品3个复孔,培养24h后,在不吸出原来培养基的条件下,空白组加入100μl新鲜无血清培养基,模型组、阳性对照组和给药组各加入100μl的100mmol/l aaph(无血清培养基),继续培养4h后。每孔加入15mg/ml的mtt溶液50μl,4h后弃去细胞上清液,每孔加入100μl dmso,振荡,使结晶物充分溶解。多功能酶标仪490nm处测量各孔的吸光度(a)值,试验重复进行3次,计算细胞存活率。
[0072]
2、对乙酰氨基酚(apap)诱导hepg2细胞肝损伤模型的建立及保肝活性筛选
[0073]
hepg2细胞株采用rpmi-1640培养基、10%胎牛血清(fbs)进行培养,置于5%co2,37℃的细胞培养箱中培养至细胞覆盖率达90%以上时用于传代。取96孔细胞培养板,每孔加入浓度为2
×
105个/ml的细胞悬液100μl,在37℃、5%co2培养箱中贴壁培养24h后,吸出原培养液弃去。设置空白组、apap模型组、阳性对照组、给药组,进行研究。空白组加100μl含10%胎牛血清的培养基,apap模型组给予100μl 7.5mmol/l浓度的apap(含10%胎牛血清的培养基溶解),阳性对照组给予100μl 2.5mg/ml异甘草酸镁的培养基(含7.5mmol/l apap),给药组给予100μl 50μmol/l化合物1和2的培养基(含7.5mmol/l apap),每个样品3个复孔,
培养48h后,每孔加入15mg/ml的mtt溶液50μl,4h后弃去细胞上清液,每孔加入100μl dmso,振荡,使结晶物充分溶解。多功能酶标仪490nm处测量各孔的吸光度(a)值,试验重复进行3次,计算细胞存活率。
[0074]
二、实验结果
[0075]
1、式1、式2所示化合物的抗氧化活性筛选结果
[0076]
表2抗氧化抑制结果
[0077][0078]
2、式1、式2所示化合物的保肝活性筛选结果
[0079]
表3抗氧化抑制结果
[0080][0081]
结论:从图中数据可以看知,化合物1和2对aaph所致肝癌细胞氧化损伤具有显著保护作用,其中化合物1的抗氧化活性优于阳性对照槲皮素;采用对乙酰氨基酚(apap)处理hepg2细胞建立体外药物性肝损伤模型,对化合物的保肝能力进行评价,化合物1和2均可不同程度抑制apap对hepg2细胞的损伤,其中化合物2的保肝活性优于化合物1,均略低于阳性对照药。
[0082]
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1