一种原位成核聚丁烯合金及其制备方法与流程
1.本发明涉及高分子材料技术领域,尤其涉及一种原位成核聚丁烯合金及其制备方法。
背景技术:
2.聚丁烯合金一般由丁烯-1和丙烯釜内聚合而成,其既具有聚丁烯-1优异的耐蠕变、耐高温、低收缩性和高抗冲性能的优点,又具备聚丙烯的高模量、高表面硬度和快速成型的特点,是一种新型的聚烯烃材料。
3.然而,聚丁烯合金的两种主要组分等规聚丁烯-1和等规聚丙烯均具有复杂的晶型结构及不同的结晶特点。等规聚丁烯-1是一种半结晶性聚合物,存在多种晶型结构,其从熔体中冷却后由热力学不稳定的晶型ii转变为稳定的晶型i周期较长,往往需要7天以上才呈现出稳定的性能;等规聚丙烯也属于半结晶性聚合物,具有结晶速率慢、结晶度较低、在成型过程中易形成较大尺寸球晶等缺点。这些结晶缺陷及问题将直接影响聚丁烯合金的综合性能及应用。
4.目前,在工业生产中主要通过在聚丁烯合金加工过程中添加成核剂的方法来加快聚合物的晶型转变过程,改善其结晶缺陷。常用的聚丁烯合金成核剂诸如芳香族酰胺类(cn 104629195 b、cn 104629184 b)、二元酸金属盐类(cn 111440387 a)、山梨醇类(cn 111286128 a)、纳米二氧化硅(cn 113563670 a)等,其添加的成核剂浓度较高,通常为500~2000ppm,如此高浓度的添加量不仅会影响聚丁烯合金的物理力学性能而且在加工过程中会出现比较难闻的气味,造成环境污染,并且成核剂无法在聚丁烯合金基体中均匀分散,影响产品质量稳定性。此外,高浓度的成核剂添加大幅增加了聚丁烯合金的加工成本。
5.聚烯烃原位成核技术是一种在聚烯烃合成的过程中直接形成聚合物成核剂的技术,该技术可以替代传统的在聚烯烃后续加工过程中添加成核剂的成核技术,由于原位成核技术中的成核剂在聚合物形成前存在于聚合体系,使得其分散效果更佳,有效改善聚合物材料的结晶性能。
6.然而,理论上,聚烯烃原位成核技术相比在聚烯烃后续加工过程中添加成核剂的成核技术具有一定优势,但是,实际生产中,如何真正达到较好的效果,仍面临一定的挑战。
技术实现要素:
7.本发明解决的技术问题在于提供一种原位成核聚丁烯合金及其制备方法,本技术提供的原位成核聚丁烯合金的制备方法可在聚合物成核剂的作用下制备原位成核聚丁烯合金,且聚丁烯合金具有高结晶度、高结晶温度和较短的晶型转变周期,实现了在较低的成核剂浓度下获得极佳结晶性能的聚丁烯合金。
8.有鉴于此,本技术提供了一种原位成核聚丁烯合金,由聚丁烯-1、α-烯烃的均聚物或共聚物以及聚合物成核剂组成;所述聚合物成核剂由主催化剂、助催化剂和聚合物成核剂单体制备得到。
9.优选的,所述聚合物成核剂的含量≤500ppm,所述聚丁烯-1的含量为80~97wt%,所述α-烯烃的均聚物或共聚物的含量为3~20wt%。
10.优选的,所述主催化剂选自非均相ziegler-natta催化剂、茂金属催化剂和后过渡金属催化剂中的一种或多种,所述助催化剂选自烷基铝化合物和有机硼化物中的一种或多种,所述聚合物成核剂单体为乙烯基化合物。
11.本技术还提供了一种原位成核聚丁烯合金的制备方法,包括以下步骤:
12.a)将主催化剂、助催化剂、聚合物成核剂单体和溶剂混合、反应,得到物料a;
13.b)将所述物料a、助催化剂和α-烯烃混合并进行α-烯烃本体聚合,再加入丁烯-1进行丁烯-1本体聚合,得到原位成核聚丁烯合金。
14.优选的,所述和α-烯烃混合的过程中,还包括外给电子体;所述外给电子体为硅烷化合物。
15.优选的,步骤a)中,所述主催化剂选自非均相ziegler-natta催化剂、茂金属催化剂和后过渡金属催化剂中的一种或多种,所述助催化剂选自烷基铝化合物和有机硼化物中的一种或多种,所述助催化剂和所述主催化剂中的活性中心金属元素的摩尔比为(1~100):1;所述聚合物成核剂单体和所述主催化剂的质量比为(0.2~5):1;所述主催化剂和所述溶剂的比例为(1~100)mg:1ml;所述反应的温度为0~50℃,时间为3~30h。
16.优选的,步骤b)中,所述助催化剂和步骤a)中主催化剂中的活性中心金属元素的摩尔比为(50~500):1;所述外给电子体和步骤a)中主催化剂中的活性中心金属元素的摩尔比为(5~30):1;所述α-烯烃和步骤a)中所述主催化剂中的活性中心金属元素的摩尔比为(1
×
104~1
×
106)∶1;所述丁烯-1和步骤a)中所述主催化剂中的活性中心金属元素的摩尔比为(1
×
104~1
×
106)∶1。
17.优选的,步骤b)中,所述α-烯烃本体聚合的温度为0~20℃,时间为3~60min;所述丁烯-1本体聚合的温度为20~80℃,时间为2~24h。
18.优选的,步骤b)中,所述丁烯-1本体聚合中通入氢气,所述氢气的分压为0.5~5bar。
19.优选的,步骤a)和步骤b)中的助催化剂均以助催化剂溶液的形式加入,所述助催化剂溶液的浓度独立的为0.5~10mol/l;所述外给电子体以外给电子体溶液的形式添加,所述外给电子体溶液的浓度为0.1~5mol/l。
20.本技术提供了一种原位成核聚丁烯合金,其由聚丁烯-1、α-烯烃的均聚物或共聚物以及聚合物成核剂组成;且聚合物成核剂由主催化剂、助催化剂和聚合物成核剂单体制备得到;所述原位成核聚丁烯合金中含有聚合物成核剂,以利于提高聚丁烯合金的结晶性能;进一步的,聚合物成核剂的浓度极低,且依旧保证聚丁烯合金的结晶性能。
21.本发明还提供了原位成核聚丁烯合金的制备方法,其首先将主催化剂、助催化剂、聚合物成核剂单体和溶剂混合反应,使体系中形成聚合物成核剂,之后引入助催化剂并加入α-烯烃,直接进行α-烯烃的本体聚合,α-烯烃聚合结束后加入丁烯-1单体进行丁烯-1本体聚合,最终制得含聚合物成核剂的原位成核聚丁烯合金,并提高了聚丁烯合金中聚丁烯-1相的结晶温度、结晶度和缩短了其晶型转变周期,实现了在较低的成核剂浓度下获得极佳结晶性能的聚丁烯合金的低成本制备。
22.实验结果表明,本发明制得的原位成核聚丁烯合金中聚合物成核剂的含量达到
500ppm以下,保持较低的成核剂浓度,同时能够使所得聚丁烯合金中聚丁烯-1相具有较高的结晶度、结晶温度和较短的晶型转变周期;具体的,聚丁烯-1相结晶温度达到80℃以上,结晶度达到55%以上,晶型转变周期缩短至95h以下。进一步的,在优选条件下,能够进一步降低成核剂浓度,具体降至320ppm以下,并且进一步提高聚丁烯合金中聚丁烯-1相的结晶度、结晶温度和缩短晶型转变周期,具体使其结晶温度提升至85℃以上,结晶度提升至57%以上,晶型转变周期缩短至70h以下。
具体实施方式
23.为了进一步理解本发明,下面结合实施例对本发明优选实施方案进行描述,但是应当理解,这些描述只是为进一步说明本发明的特征和优点,而不是对本发明权利要求的限制。
24.鉴于现有技术中,原位成核聚丁烯合金的性能需求,本技术提供了一种原位成核聚丁烯合金,其在低浓度聚合物成核剂的基础上仍然实现了聚丁烯合金的高结晶度、高结晶温度、较短的晶型转变周期,最终使得聚丁烯合金具有极佳的结晶性能。具体的,本发明实施例公开了一种原位成核聚丁烯合金,由聚丁烯-1、α-烯烃的均聚物或共聚物以及聚合物成核剂组成;所述聚合物成核剂由主催化剂、助催化剂和聚合物成核剂单体制备得到。
25.在所述原位成核聚丁烯合金中,所述聚合物成核剂的含量≤500ppm,所述聚丁烯-1的含量为80~97wt%,所述α-烯烃的均聚物或共聚物的含量为3~20wt%。所述聚合物成核剂并非是直接加入的,而是在聚合反应过程中生成的。
26.本发明还提供一种原位成核聚丁烯合金的制备方法,包括以下步骤:
27.a)将主催化剂、助催化剂、聚合物成核剂单体和溶剂混合、反应,得到物料a;
28.b)将所述物料a、助催化剂和α-烯烃混合并进行α-烯烃本体聚合,再加入丁烯-1进行丁烯-1本体聚合,得到原位成核聚丁烯合金。
29.在本技术中,所述α-烯烃选自乙烯、丙烯、己烯、辛烯中的一种或几种,优选乙烯和/或丙烯,进一步优选丙烯。
30.在原位成核聚丁烯合金的制备过程中,首先将主催化剂、助催化剂、聚合物成核剂单体和溶剂混合反应,使体系中形成聚合物成核剂并包括未活化的主催化剂,之后引入助催化剂并加入α-烯烃,直接进行α-烯烃的本体聚合,α-烯烃聚合结束后可选的脱除溶剂及未反应单体,最后加入丁烯-1单体并通入氢气升温进行丁烯-1本体聚合,最终制得含低浓度聚合物成核剂的原位成核聚丁烯合金,并提高了聚丁烯合金中聚丁烯-1相的结晶温度、结晶度和缩短了其晶型转变周期,使得聚丁烯合金在较低成核剂浓度下呈现出极佳的结晶性能,在工业和商业中具有重要意义。
31.更具体的,本技术所述原位成核聚丁烯合金的制备方法,包括以下步骤:
32.a)将主催化剂、助催化剂、聚合成核剂单体和溶剂混合反应,得到物料a;
33.b)将所述物料a与助催化剂、外给电子体混合,并加入α-烯烃单体进行α-烯烃本体聚合;
34.c)任选的脱除溶剂及未反应的α-烯烃单体;
35.d)加入液相丁烯-1并通入氢气升温进行丁烯-1本体聚合,得到原位成核聚丁烯合金。
36.本发明中,所述步骤a)~c)优选在保护性气氛下进行,具体的,在投料之前,预先与反应装置进行气体置换,使反应装置中形成保护性气氛,然后再进行步骤a)~c)的投料和反应。本发明对形成保护性气氛围的气体种类没有特殊限制,为本领域常规惰性气体即可,如氮气、氦气或氩气等。
37.对于步骤a):将主催化剂、助催化剂、聚合物成核剂单体和溶剂混合反应,得到物料a。
38.本发明中,所述主催化剂选自非均相ziegler-natta催化剂、茂金属催化剂和后过渡金属催化剂中的一种或多种,具体选自ziegler-natta催化剂,更具体为氯化镁负载四氯化钛球形催化剂。本发明对所述主催化剂的来源没有特殊限制,为市售商业品即可,例如在本发明的一些实施例中,采用辽宁向阳科化集团催化剂公司的cs系列催化剂,钛含量为2.3wt%。
39.本发明中,所述助催化剂选自烷基铝化合物和有机硼化物中的一种或多种,具体为烷基铝化合物,更具体选自三乙基铝、三异丁基铝、三己基铝、三辛基铝、二乙基一氯化铝、二异丁基一氯化铝、二己基一氯化铝和二辛基一氯化铝中的至少一种;更优选为以上烷基铝化合物中的一种或两种。
40.本发明中,所述助催化剂优选以助催化剂溶液的形式添加,即预先将助催化剂溶于溶剂中配制成助催化剂溶液,然后将该助催化剂溶液添加进反应体系中。其中,配制助催化剂溶液所用的溶剂优选为正己烷、正庚烷和正辛烷中的至少一种。所述助催化剂溶液的浓度优选为0.5~8mol/l,更优选为1mol/l。
41.本发明中,所述聚合物成核剂单体为用于形成聚合物成核剂的单体,为乙烯基化合物;优选为乙烯基环己烷、乙烯基环戊烷、乙烯基-2-甲基环己烷、3-甲基-1-丁烯、3-乙基-1-己烯、3-甲基-1-戊烯和苯乙烯中的至少一种。
42.本发明中,所述溶剂优选为烷烃溶剂,更优选为正戊烷、异戊烷、正已烷、正庚烷和正辛烷中的至少一种。
43.本发明中,步骤a)中,各物料之间的用量关系优选如下:
44.所述助催化剂∶主催化剂中活性中心金属元素的摩尔比优选为(1~100)∶1,具体可为1∶1、10∶1、20∶1、30∶1、40∶1、50∶1、60∶1、70∶1、80∶1、90∶1或100∶1。
45.所述聚合物成核剂单体∶主催化剂的质量比为(0.2~5)∶1,具体可为0.2∶1、0.5∶1、1.0∶1、1.5∶1、2.0∶1、2.5∶1、3.0∶1、3.5∶1、4.0∶1、4.5∶1、5.0∶1,控制在以上比例范围下,能够使聚丁烯合金中聚合物成核剂的含量达到500ppm以下,保持较低的成核剂浓度,同时能够使所得聚丁烯合金中的聚丁烯-1相具有较高的结晶度、结晶温度和较短的晶型转变周期,具体的,产物结晶温度达到80℃以上,结晶度达到55%以上,晶型转变周期缩短至95h以下。本发明中,所述聚合物成核剂单体∶主催化剂的质量比更优选为(2~5)∶1,在该优选比例下,能够进一步提高聚丁烯合金中聚丁烯-1相的结晶度、结晶温度和缩短晶型转变周期,具体的,其结晶温度提升至85℃以上,结晶度提升至57%以上,晶型转变周期缩短至70h以下。
46.所述主催化剂∶溶剂的用量比优选为(1~100)mg∶1ml。
47.本发明中,将主催化剂、助催化剂、聚合物成核剂单体和溶剂这4种物料混合后进行反应,得到物料a。上述反应为聚合反应,在反应过程中,以氧化镁负载四氯化钛球形催化
剂为例,助催化剂将球形主催化剂表面负载的四氯化钛还原为三氯化钛或二氯化钛等低价钛,低价钛作为活性中心引发聚合物成核剂单体在主催化剂球形表面进行聚合反应生成聚合物成核剂,并均匀分布在球形主催化剂表面;该反应中仅有小部分主催化剂参与聚合物成核剂的制备,绝大部分催化剂未被活化,而继续参与后期的烯烃聚合,因此物料a为主催化剂和聚合物成核剂的混合物。所述反应的温度优选为0~50℃,具体可为0℃、5℃、10℃、15℃、20℃、25℃、30℃、35℃、40℃、45℃、50℃。所述反应的时间优选为3~30h,具体可为3h、5h、10h、15h、20h、25h、30h。经上述反应后,得到物料a。
48.对于步骤b):将所述物料a与助催化剂、外给电子体混合,并加入α-烯烃单体进行本体预聚合。
49.上述过程中,所述助催化剂选自烷基铝化合物和有机硼化物中的一种或多种,具体为烷基铝化合物,优选为三乙基铝、三异丁基铝、三己基铝、三辛基铝、二乙基一氯化铝、二异丁基一氯化铝、二己基一氯化铝和二辛基一氯化铝中的至少一种;更优选为以上烷基铝化合物中的一种或两种。所述助催化剂与步骤a)中采用的助催化剂可以相同或不同,优选为相同。
50.本发明中,所述助催化剂优选以助催化剂溶液的形式添加,即预先将助催化剂溶于溶剂中配制成助催化剂溶液,然后将该助催化剂溶液添加进反应体系中。其中,配制溶液所用的溶剂优选为正己烷、正庚烷和正辛烷中的至少一种。所述助催化剂溶液的浓度优选为0.5~8mol/l,更优选为1mol/l。
51.本发明中,所述助催化剂∶步骤a)主催化剂中活性中心金属元素的摩尔比优选为(50~500)∶1,具体可为50∶1、100∶1、150∶1、200∶1、250∶1、300∶1、350∶1、400∶1、450∶1或500∶1。
52.本发明中,所述外给电子体为硅烷化合物,优选为乙烯基三乙氧基硅烷、乙烯基三甲氧基硅烷、甲基三乙氧基硅烷、二甲基二甲氧基硅烷、二乙基二甲氧基硅烷、二丙基二甲氧基硅烷、二异丙基二甲氧基硅烷、二丁基二甲氧基硅烷、二异丁基二甲氧基硅烷、二叔丁基二甲氧基硅烷、二叔己基二甲氧基硅烷、二苯基二甲氧基硅烷、二环己基二甲氧基硅烷、二环戊基二甲氧基硅烷、二甲基二乙氧基硅烷、二乙基二乙氧基硅烷、二丙基二乙氧基硅烷、二异丙基二乙氧基硅烷、二丁基二乙氧基硅烷、二异丁基二乙氧基硅烷、二叔丁基二乙氧基硅烷、二叔己基二乙氧基硅烷、二苯基二乙氧基硅烷、二环己基二乙氧基硅烷、二环戊基二乙氧基硅烷、苯基三甲氧基硅烷、苯基三乙氧基硅烷、环戊基甲基二甲氧基硅烷、环戊基三甲氧基硅烷、环己基甲基二甲氧基硅烷、环己基三甲氧基硅烷、叔己基三甲氧基硅烷、叔丁基三甲氧基硅烷和叔己基三甲氧基硅烷中的至少一种;更优选为以上硅烷化合物中的一种或两种。
53.本发明中,所述外给电子体优选以外给电子体溶液的形式添加,即预先将外给电子体溶于溶剂中配制成外给电子体溶液,然后将该外给电子体溶液添加进反应体系中。其中,配制外给电子体溶液所用的溶剂优选为正戊烷、异戊烷、正已烷和正更烷中的至少一种。所述外给电子体溶液的浓度优选为0.1~5mol/l,更优选为0.2mol/l。
54.本发明中,所述外给电子体∶步骤a)主催化剂中活性中心金属元素的摩尔比优选为(5~30)∶1,具体可为5∶1、10∶1、15∶1、20∶1、25∶1或30∶1。
55.本发明中,除以上物料外,向体系中加入α-烯烃单体,选自乙烯、丙烯、己烯、辛烯
中的一种或多种。本发明中,所述α-烯烃单体:步骤a)主催化剂中活性中心金属元素的摩尔比优选为(1
×
104~1
×
106)∶1,具体可为1
×
104∶1、1
×
105∶1或1
×
106∶1。
56.在上述过程中,上述物料的混合优选在低温条件下进行,即预先将体系降至低温条件,再投加物料。所述低温条件是指温度≤20℃,更优选为0℃~20℃,具体可为0℃、5℃、10℃、15℃、20℃。
57.本发明中,将以上所有物料加完后,控制在低温条件下进行α-烯烃本体聚合。本发明中,所述低温条件是指温度≤20℃,更优选为0℃~20℃,具体可为0℃、5℃、10℃、15℃或20℃。控制在上述低温条件下进行α-烯烃本体聚合的时间优选为3~60min,具体可为3min、5min、10min、15min、20min、30min、40min、50min或60min。控制在以上α-烯烃本体聚合时间范围下,能够使聚丁烯合金中聚合物成核剂的含量达到500ppm以下,并且保证聚丁烯合金具有优异的结晶性能。本发明中,所述α-烯烃本体聚合时间更优选为5~15min,在该优选比例下,能够进一步降低成核剂浓度,具体降至320ppm以下,并且保证使所得聚丁烯合金中聚丁烯-1相具有较高的结晶度、结晶温度和较短的晶型转变周期。
58.经上述α-烯烃本体聚合后,得到α-烯烃聚合物与溶剂及未反应丙烯单体的共混物。
59.对于步骤c):任选的脱除溶剂及未反应的α-烯烃单体。
60.本发明中,在上述丁烯-1本体聚合反应前,对步骤b)所得共混物体系任选的脱除溶剂及未反应α-烯烃单体。本发明中,所述脱除溶剂及未反应α-烯烃单体的方式优选为:利用隔膜真空泵对反应体系抽真空将溶剂及未反应丙烯单体抽除分离。上述操作的温度条件优选为30~60℃。
61.对于步骤d):加入液相丁烯-1并通入氢气升温进行丁烯-1本体聚合,得到原位成核聚丁烯合金。
62.本发明中,本发明中,所述丁烯-1单体:步骤a)主催化剂中活性中心金属元素的摩尔比优选为(1
×
104~1
×
106)∶1,具体可为1
×
104∶1、1
×
105∶1或1
×
106∶1。
63.在上述过程中还向体系中通入氢气。本发明中,所述氢气的分压优选为0.5~5bar,具体可为0.5bar、1bar、1.5bar、2bar、2.5bar、3bar、3.5bar、4bar、4.5bar或5bar。
64.本发明中,加入丁烯-1并通入氢气后继续升温进行丁烯-1本体聚合。本发明中,所述本体聚合的温度优选为20~60℃,具体可为20℃、25℃、30℃、35℃、40℃、45℃、50℃、55℃或60℃。本发明中,步骤c)是进一步升温进行丁烯-1本体聚合,即丁烯-1本体聚合的温度>步骤b)中丙烯本体聚合的温度,隐含了步骤d)本体聚合的温度与步骤b)本体预聚合的温度不同时为20℃。本发明中,所述丁烯-1本体聚合的时间优选为2~20h,具体可为2h、4h、6h、8h、10h、12h、14h、16h、18h或20h。经上述丁烯-1本体聚合后,体系中生成原位成核聚丁烯合金。
65.本发明中,在上述丁烯-1本体聚合后,优选对所得反应液进行如下后处理:将未反应的丁烯-1单体通过排气阀通入丁烯-1回收罐,精制回收循环使用。经上述后处理后,得到原位成核聚丁烯合金。
66.本发明提供了一种原位成核聚丁烯合金的制备方法,其首先将主催化剂、助催化剂、聚合物成核剂单体和溶剂混合反应,使体系中形成聚合物成核剂,之后引入助催化剂并加入α-烯烃单体,直接进行α-烯烃的本体聚合,α-烯烃聚合结束后可选的脱除溶剂及未反
应α-烯烃单体,加入丁烯-1单体并通入氢气升温进行丁烯-1本体聚合,最终制得含低浓度聚合物成核剂的原位成核聚丁烯合金,并提高了聚丁烯合金中聚丁烯-1相的结晶温度、结晶度和缩短了其晶型转变周期,实现了在较低的成核剂浓度下获得极佳结晶性能的聚丁烯合金的低成本制备。
67.实验结果表明,本发明制备的原位成核聚丁烯合金中聚合物成核剂的含量达到500ppm以下,保持较低的成核剂浓度,同时能够使所得聚丁烯合金中聚丁烯-1相具有较高的结晶度、结晶温度和较短的晶型转变周期,具体的,聚丁烯-1相结晶温度达到80℃以上,结晶度达到55%以上,晶型转变周期缩短至95h以下。进一步的,在优选条件下,能够进一步降低成核剂浓度,具体降至320ppm以下,并且进一步提高聚丁烯合金中聚丁烯-1相的结晶度、结晶温度和缩短晶型转变周期,具体使其结晶温度提升至85℃以上,结晶度提升至57%以上,晶型转变周期缩短至70h以下。
68.为了进一步理解本发明,下面结合实施例对本发明提供的原位成核聚丁烯合金及其制备方法进行详细说明,本发明的保护范围不受以下实施例的限制。
69.以下实施例和对比例中,所用主催化剂为市售氯化镁负载四氯化钛型,辽宁向阳科化集团催化剂公司的cs系列,钛含量为2.3wt%;反应装置为3l高压聚合釜,其搅拌桨与聚合釜底间距1~3mm;步骤s1中所用溶剂为精制处理去除水分和杂质的溶剂。
70.实施例1
71.s1、3升的高压釜用高纯氮气充分置换后,依次加入100mg主催化剂、0.5ml助催化剂溶液(助催化剂为三乙基铝,溶剂为正己烷,溶液浓度为1mol/l)、100mg乙烯基环己烷(即vch,密度0.805g/ml,纯度≥98%,水含量≤100ppm)、100ml正己烷,在30℃聚合12h,得到物料a;
72.s2、将釜温降至20℃,加入5ml助催化剂溶液(助催化剂为三乙基铝,溶剂为正己烷,溶液浓度为1mol/l)、5ml外给电子体溶液(外给电子体为二异丁基二甲氧基硅烷,溶剂为正庚烷,溶液浓度为0.2mol/l),然后,再加入80g液相丙烯,在20℃下进行丙烯本体预聚合10min;
73.s3、丙烯本体聚合结束后,保持在30℃下用隔膜真空泵对反应釜抽真空将溶剂及未反应丙烯单体抽出分离,然后,加入800g液相丁烯-1和1bar氢气,将反应釜升温至40℃,继续聚合3h,得到原位成核聚丁烯合金。
74.实施例2
75.按照实施例1实施,区别在于:vch用量调整为200mg。
76.实施例3
77.按照实施例1实施,区别在于:vch用量调整为300mg。
78.实施例4
79.按照实施例1实施,区别在于:vch用量调整为500mg,vch聚合时间为24h
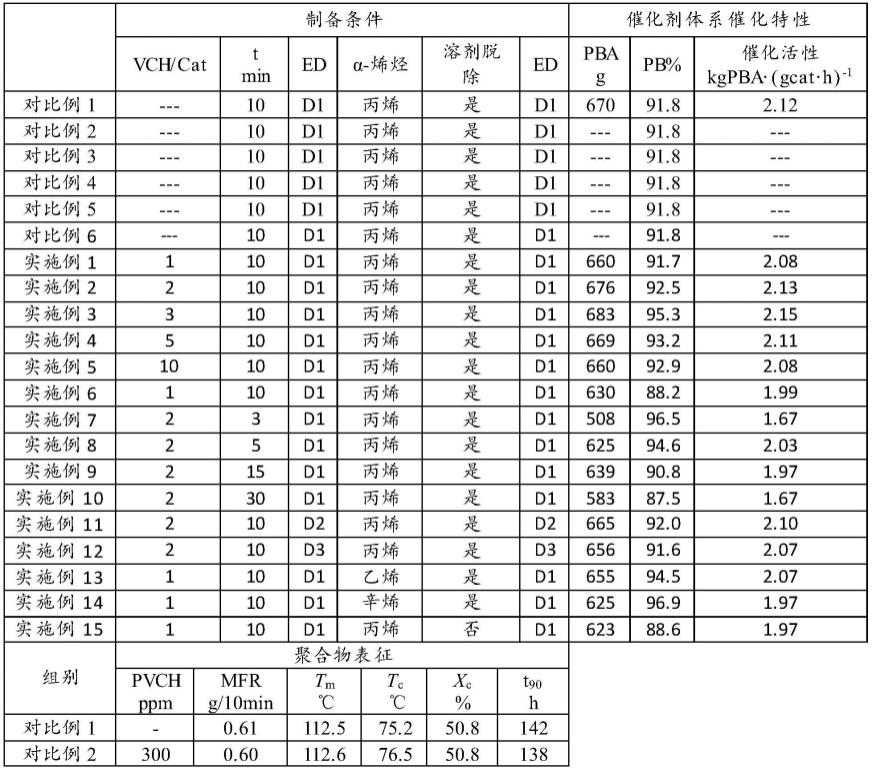
80.实施例5
81.按照实施例1实施,区别在于:vch用量调整为1000mg,vch聚合时间为30h。
82.实施例6
83.按照实施例1实施,区别在于:vch替换为3-甲基-1-丁烯。
84.实施例7
85.按照实施例2实施,区别在于:丙烯本体聚合时间为3min。
86.实施例8
87.按照实施例2实施,区别在于:丙烯本体聚合时间为5min。
88.实施例9
89.按照实施例2实施,区别在于:丙烯本体聚合时间为15min。
90.实施例10
91.按照实施例2实施,区别在于:丙烯本体聚合时间为30min。
92.实施例11
93.按照实施例2实施,区别在于:外给电子体选用二环己基二甲氧基硅烷。
94.实施例12
95.按照实施例2实施,区别在于:外给电子体选用二环戊基二甲氧基硅烷。
96.实施例13
97.按照实施例1实施,区别在于:丙烯替换为乙烯。
98.实施例14
99.按照实施例1实施,区别在于:丙烯替换为辛烯
100.实施例15
101.按照实施例1实施,区别在于:丙烯本体聚合结束后,溶剂和未反应的丙烯单体不进行分离,直接加入丁烯-1。
102.对比例1
103.3升的高压釜用高纯氮气充分置换后,依次加入100mg主催化剂、5ml助催化剂溶液(助催化剂为三乙基铝,溶剂为正己烷,溶液浓度为1mol/l)、5ml外给电子体溶液(外给电子体为二异丁基二甲氧基硅烷,溶剂为正庚烷,溶液浓度为0.2mol/l),然后在加入80g液相丙烯,在20℃下进行丙烯本体预聚合10min;丙烯本体聚合结束后,保持在30℃下用隔膜真空泵对反应釜抽真空将溶剂及未反应丙烯单体抽出分离,然后,再加入800g液相丁烯-1和1bar氢气,将反应釜升温至40℃,继续聚合3h,得到聚丁烯合金。
104.对比例2
105.s1、500ml的高压釜用高纯氮气充分置换后,依次加入100mg主催化剂、0.5ml助催化剂溶液(助催化剂为三乙基铝,溶剂为正己烷,溶液浓度为1mol/l)、1000mg乙烯基环己烷(即vch,密度0.805g/ml,纯度≥98%,水含量≤100ppm)、100ml正己烷,在30℃聚合30h,得到物料a,干燥后获得含催化剂体系的聚合物成核剂(pvch);
106.s2、按照对比例1制备的聚丁烯合金150g与按照对比例2的s1制备的50mgpvch物理共混。
107.对比例3
108.按照对比例2实施,区别在于:pvch用量为73mg。
109.对比例4
110.按照对比例2实施,区别在于:,pvch用量为124mg。
111.对比例5
112.按照对比例2实施,区别在于:pvch用量为248mg。
113.对比例6
114.按照对比例2实施,区别在于:pvch用量为330mg。
115.将实施例和对比例制备的聚丁烯合金进行性能检测,具体为:
116.1、聚合物中聚乙烯基环己烷(pvch)含量的测试:通过测试乙烯基环己烷(vch)的转化率计算;另外,用气相色谱仪分析步骤s1中聚合结束后未分离溶剂前的反应液中残留vch的量;甲苯用作内标;以上实施例和对比例中vch残余量均为0;
117.2、熔体质量流动速率(mfr)的测试:按照gb/t3682.1-2018测定,负荷2.16kg,测试温度190℃;
118.3、合金中聚丁烯-1质量分数(pb%)的测试:采用乙醚和正庚烷二段抽提法;首先,将2g干燥的聚合物样品,放在索氏抽提器中用沸腾乙醚抽提48h后,将乙醚不溶物干燥后放在索氏抽提器中用沸腾正庚烷抽提48h,将正庚烷可溶物干燥至恒重所得到的聚合物质量(g)与2(g)的比值即为合金中聚丁烯-1的质量分数;
119.4、dsc测试:在氮气气氛,首先以10℃/min的速率将5~10mg样品从室温加热到200℃,并在200℃下保持5min以消除热历史;然后,以-10℃/min的速率将样品冷却至室温;最后,以10℃/min的速率将样品重新加热至200℃;根据以下关系式,使用第二次升温曲线计算样品的结晶温度(tc)和结晶度(xc),熔点(tm)也根据二次升温曲线测得:xc(%)=δhm/(δh
*f
·
pb%)
·
100%,其中,δhm是二次升温曲线聚丁烯-1相的熔融焓,δh
*f
是聚丁烯-1晶型ii的标准熔融焓(62j/g),pb%为体系中聚丁烯-1的质量分数;
120.5、催化活性的测试:按以下公式计算ca=q/w
cat
·
t
×
10-3
,单位为kgpba
·
(gcat
·
h)-1
,其中ca为催化剂催化活性,q为在聚合反应产物质量(g),w
cat
为主催化剂用量(g),t为丙烯和丁烯-1本体聚合时间之和(h);
121.6、晶型转变周期(t
90
)的测试:晶型检测采用的是广角x射线衍射(waxd),测试采用cu靶作为阳极金属材料,产生的波长为0.154nm的kα线,测试电压为40kv,电流为40ma,扫描速率为5
°
/min,扫描范围为5
°‑
40
°
;测试样品以-50℃/min的速度从200℃快速降温至25℃,在25℃对不同退火时间的样品进行表征;waxs测试结果中,在11.9
°
、16.9
°
和18.5
°
观察到的衍射峰可分别归属于聚丁烯-1中晶型ii的(200)、(220)和(213)晶面;在10.0
°
、17.5
°
和20.4
°
处观察到的衍射峰可分别归属于聚丁烯-1晶型i的(110)、(300)和(220/+211)晶面,i(110)i和i(200)
ii
分别代表晶型i(110)晶面和晶型ii(200)晶面衍射峰的积分强度,使用i(110)i/[i(110)i+0.67i(200)
ii
]表示晶型i完成转变的百分比(xi),t
90
即聚丁烯-1熔融冷却后晶型i完成90%转变(xi=90)的时间。
[0122]
测试结果参见表1:
[0123]
表1实施例和对比例聚丁烯合金的制备及聚合物表征结果数据表
[0124][0125][0126]
结晶温度是成核剂效率的良好指标,较高的结晶温度意味着最终产品中更有效的成核。由表1测试结果可以看出:与对比例1相比,实施例1~15均呈现出较好的成核效果,使聚丁烯-1相的结晶温度由75℃提高到了80℃以上,且结晶度由50%提高到了55%以上,晶型转变周期缩短至95h以下。实施例1~15中,实施例2~5、7~12处于优选条件下,使所得聚
丁烯-1合金中聚丁烯-1相的结晶温度、结晶度和晶型转变周期进一步显著改善,具体使其结晶温度提升至85℃以上,结晶度提升至57%以上,晶型转变周期缩短至70h以下。
[0127]
实施例2~5与对比例2~5比较,在相同的聚合物成核剂浓度下,原位成核技术相比于现有技术对聚丁烯合金的结晶性能改善效果更加,传统加工成核pvch浓度为2000ppm时与该发明技术的300ppm结晶性能相当,说明该发明技术可以大大降低成核剂浓度,且能保证聚丁烯-1良好的结晶性能。
[0128]
以上实施例的说明只是用于帮助理解本发明的方法及其核心思想。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明权利要求的保护范围内。
[0129]
对所公开的实施例的上述说明,使本领域专业技术人员能够实现或使用本发明。对这些实施例的多种修改对本领域的专业技术人员来说将是显而易见的,本文中所定义的一般原理可以在不脱离本发明的精神或范围的情况下,在其它实施例中实现。因此,本发明将不会被限制于本文所示的这些实施例,而是要符合与本文所公开的原理和新颖特点相一致的最宽的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1