制备3,4-二氢喹唑啉衍生物的方法与流程
1.本发明属于药物合成领域,具体涉及一种制备3,4-二氢喹唑啉衍生物的方法。
背景技术:
2.3,4-二氢喹唑啉衍生物(4s)-2-{8-氟-2-[4-(3-甲氧基苯基)哌嗪-1-基]-3-[2-甲氧基-5(三氟甲基)苯基]-3,4-二氢喹唑啉-4-基}乙酸是一种新型抗巨细胞病毒药,具有如下式i所示结构。目前正开发用于预防cmv感染,用于接受异基因造血干细胞移植(hsct)后cmv血清呈阳性的成人患者预防cmv感染和相关疾病。
[0003][0004]
现有技术wo2015088931a1公开了式i化合物的制备方法,采用2-溴-6-氟苯胺为其原料,经heck偶联、缩合、成脲、成亚胺、成胍、环合、拆分、水解等步骤制备。
技术实现要素:
[0005]
一方面,本发明提供了一种分离(s)-2-{8-氟-2-[4-(3-甲氧基苯基)哌嗪-1-基]-3-[2-甲氧基-5-(三氟甲基)苯基]-3,4-二氢喹唑啉-4-基}乙酸(c1-c4)烷基酯的方法,将2-{8-氟-2-[4-(3-甲氧基苯基)哌嗪-1-基]-3-[2-甲氧基-5-(三氟甲基)苯基]-3,4-二氢喹唑啉-4-基}乙酸(c1-c4)烷基酯与(2s,3s)-二对甲基苯甲酰酒石酸在丙酸甲酯中结晶,s-对映体的盐首先从溶液中沉淀出来,反应路线如下:
[0006][0007]
其中,n选自0、1、2或3。
[0008]
在一些实施方案中,n选自0、1或2;在一些典型的实施方案中,n选自0或1;在一些更典型的实施方案中,n为1。
[0009]
在一些实施方案中,所述反应在0~30℃进行;优选的,所述反应在0~20℃进行;
更优选的,所述反应在5~10℃进行。
[0010]
另一方面,本发明提供了一种制备中间体int7的方法,包括以下步骤:
[0011][0012]
其中,n选自0、1、2或3;
[0013]
将化合物int5在催化剂存在下于有机溶剂丙酸甲酯中发生环合反应生成化合物int6,反应完成后,将反应体系降温,加入(2s,3s)-二对甲基苯甲酰酒石酸进行结晶反应,分离得到中间体int7。
[0014]
在一些实施方案中,n选自0、1或2;优选的,n选自0或1;更优选的,n为1。
[0015]
在一些实施方案中,所述催化剂选自(r,r)-n,n'-双(三氟甲烷磺酰)-1,2-二苯基乙二胺,(r,r)-n,n'-双(三氟甲烷磺酰)-1,2-二吡啶基乙二胺,(r,r)-n,n'-双(三氟甲烷磺酰)-1,2-二(3-氰基苯基)乙二胺或(r,r)-n,n'-双(三氟甲烷磺酰)-1,2-二(3-甲氧基苯基)乙二胺;在一些典型的实施方案中,所述催化剂选自(r,r)-n,n'-双(三氟甲烷磺酰)-1,2-二苯基乙二胺或(r,r)-n,n'-双(三氟甲烷磺酰)-1,2-二吡啶基乙二胺;在一些更典型的实施方案中,所述催化剂选自(r,r)-n,n'-双(三氟甲烷磺酰)-1,2-二苯基乙二胺。
[0016]
在一些实施方案中,所述环合反应的反应温度为50~80℃;优选的,所述环合反应的反应温度为55~80℃;更优选的,所述环合反应的温度为60~80℃。
[0017]
在一些实施方案中,将反应体系降温至-5~25℃;优选的,将反应体系降温至-5~15℃;更优选的,将反应体系降温至-5~5℃。
[0018]
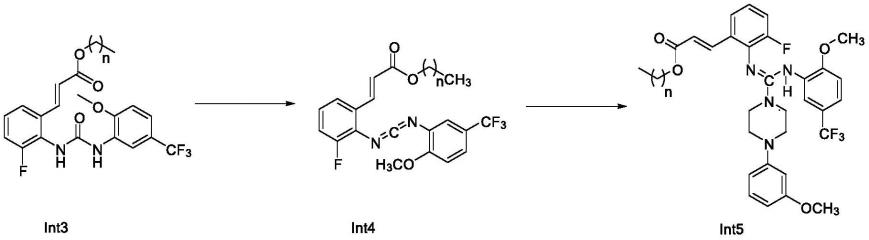
在一些实施方案中,中间体int5的制备方法包括以下步骤:
[0019][0020]
其中,n具有如上文所述定义;
[0021]
将化合物int3、碱以及含磷试剂在有机溶剂中发生成亚胺反应,生成中间体int4,中间体int4与1-(3-甲氧基苯基)哌嗪或其盐发生成胍反应,生成中间体int5。
[0022]
在一些实施方案中,所述成亚胺反应的有机溶剂选自甲苯、二氯甲烷或四氢呋喃。
[0023]
在一些实施方案中,所述成亚胺反应采用的碱选自2-甲基吡啶、吡啶、3-甲基吡啶或4-甲基吡啶;优选2-甲基吡啶。
[0024]
在一些实施方案中,含磷试剂选自五氯化磷或三氯氧磷;优选五氯化磷。
[0025]
在一些实施方案中,所述成胍反应的有机溶剂选自甲苯、二氯甲烷或四氢呋喃。
[0026]
在一些实施方案中,中间体int4与1-(3-甲氧基苯基)哌嗪二盐酸盐发生成胍反应。
[0027]
在一些实施方案中,中间体int4不经分离,直接进行下一步成胍反应。
[0028]
在一些实施方案中,成胍反应在碱性条件下进行,优选的碱性试剂为三乙胺。
[0029]
在一些实施方案中,所述中间体int3的制备方法包括以下步骤:
[0030][0031]
其中,n具有如上文所述定义;
[0032]
将化合物int2与2-甲氧基-5-三氟甲基苯胺于有机溶剂中发生成脲反应,生成中间体int3。
[0033]
在一些实施方案中,所述成脲反应在催化剂存在条件下进行,所述催化剂优选二甲基氨基吡啶。
[0034]
在一些实施方案中,所述成脲反应的有机溶剂选自乙酸异丙酯、乙酸乙酯、甲苯或dmf;优选乙酸异丙酯。
[0035]
在一些实施方案中,所述中间体int2的制备方法包括以下步骤:
[0036][0037]
其中,n具有如上文所述定义;
[0038]
将原料sm1与原料sm2在醋酸钯催化体系和碱的条件下于有机溶剂中发生偶联反应,生成中间体int1;中间体int1与氯甲酸苯酯或溴甲酸苯酯发生缩合反应生成中间体int2。
[0039]
在一些实施方案中,所述醋酸钯催化体系的配体选自三(邻甲苯基)膦。
[0040]
在一些实施方案中,所述偶联反应的碱选自n,n-二异丙基乙胺、三乙胺、n-甲基二环己基胺或n-甲基-二异丙基乙基胺。
[0041]
在一些实施方案中,所述偶联反应的有机溶剂选自n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、甲苯、乙酸异丙酯或乙酸乙酯。在一些实施方案中,所述缩合反应在选自n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、甲苯、乙酸异丙酯或乙酸乙酯的溶剂中进行。
[0042]
在一些实施方案中,所述缩合反应在选自磷酸氢二钠、碳酸氢钠、吡啶、三乙胺或二异丙基乙基胺的碱性条件下反应。
[0043]
在一些实施方案中,所述偶联反应生成的中间体int1不经分离,直接进行下一步
缩合反应。
[0044]
另一方面,本发明还提供了由中间体int7进一步制备终产品化合物i的方法,包括将中间体int7先游离再发生水解反应制备得到化合物i。
[0045]
在一些实施方案中,所述游离步骤在碱性试剂存在下进行,所述碱性试剂选自碳酸氢钠、碳酸氢钾、吡啶、三乙胺或二异丙基乙基胺的碱性条件下进行。
[0046]
在一些实施方案中,所述游离步骤在选自乙酸乙酯、乙酸异丙酯、甲苯或二氯甲烷的溶剂中进行。
[0047]
在一些实施方案中,所述水解反应在选自氢氧化锂、氢氧化钠或氢氧化钾的碱性水溶液条件下进行。
[0048]
在一些实施方案中,所述水解反应在选自丙酮、1,4-二氧六环、四氢呋喃、乙醇或甲醇的溶剂中进行。
[0049]
有益效果:本发明提供的制备化合物i的中间体int7的方法采用丙酸甲酯作为环合和拆分步骤的溶剂,减少了溶剂种类,且环合步骤原料转化率高,环合步骤结束后直接将体系降温后加入酒石酸成盐,避免了浓缩的步骤,简化了工艺,适合工业上放大使用。
[0050]
相关定义:
[0051]
dmf:n,n-二甲基甲酰胺
[0052]
dcm:二氯甲烷
[0053]
ea:乙酸乙酯
[0054]
tea:三乙胺
[0055]
dmap:4-二甲氨基吡啶
具体实施方式
[0056]
下面结合优选实施例对本发明作进一步说明,下述实施例仅用于说明本发明而并非对本发明的限制。
[0057]
hplc分析方法:
[0058]
检测方法a:
[0059]
柱:ymc-pack ods-a(4.6*250mm,5μm)
[0060]
溶剂:乙腈
[0061]
流动相:0.1%甲酸溶液:乙腈(70:30)
[0062]
检测波长:220nm
[0063]
柱温:30℃
[0064]
进样体积:10μl
[0065]
检测方法b:
[0066]
柱:chiralpak ad-h(4.6*250mm,5μm)
[0067]
溶剂:流动相
[0068]
流动相:正己烷-乙醇(80:20)
[0069]
检测波长:254nm
[0070]
柱温:30℃
[0071]
进样体积:10μl
[0072]
实施例1环合步骤溶剂的考察
[0073]
分别取4份int5(1.0g)和催化剂((r,r)-n,n'-双(三氟甲烷磺酰)-1,2-二苯基乙二胺,40mg),依次编号w1、w2、w3和w4。分别向w1、w2、w3、w4中加入乙酸异丙酯(5ml)、dmf(5ml)、etoh(5ml)和丙酸甲酯(5ml)。升至55℃反应29.5h。取样检测有关物料(检测方法a)和异构体(检测方法b)。
[0074]
结果:w1中原料剩余11.14%,产物中r异构体8.9%;w2中原料剩余79.38%,未检测r异构体;w3中原料tlc点板基本无剩余,产物中r异构体48.5%;w4中原料剩余2.72%,产物中r异构体9.69%。
[0075]
实施例2偶联反应步骤催化剂的考察
[0076]
分别将5份2-溴-6-氟苯胺(1.0g)、丙烯酸乙酯(1.05g)和n,n-二环己基甲基胺(1.2g)加至dmf(3ml)中,反应1中加入1.5当量(dba)2pd,反应2中加入1.5当量(pph3)4pd,反应3中加入1.5当量pd(oac)2,反应4中加入1.5当量双(二叔丁基-4-二甲氨基苯基膦)氯化钯,反应5中加入1.5当量pd-g2。氮气置换后升至100℃反应2h。取样检测(检测方法a)。结果如下:
[0077] 反应1反应2反应3反应4反应5原料剩余(%)54.541.32.72.10.06杂质(%)未检出未检出n/a0.030.17
[0078]
实施例3制备中间体int2(n=1)
[0079]
将500g 2-溴-6-氟苯胺加入1500ml dmf中,搅拌下加入525g丙烯酸乙酯和410g dipea,氮气置换。加入56g三(邻甲基苯基)膦,加6g醋酸钯,氮气置换。75~85℃,保温反应1h。反应液过滤,滤饼用乙酸异丙酯淋洗,合并滤液。滤液用纯化水洗涤一次。向所得反应液中加入560.40g磷酸氢二钠与2500ml水配置的溶液,15~25℃搅拌反应,后滴加515.00g氯甲酸苯酯,反应过夜。
[0080]
反应液过滤,滤饼用水洗涤。静置分液,取有机相,有机相中加入7000ml正庚烷,打浆,过滤,滤饼正庚烷洗。所得固体45℃鼓风干燥,得类白色固体614g,收率70.8%。
[0081]
实施例4制备中间体int3(n=1)
[0082]
将426g int2、285.4g 2-甲氧基-5-(三氟甲基)苯胺和7.9gdmap加至4400ml乙酸异丙酯中,升温至80℃,反应3h。
[0083]
反应完毕,降温至36℃,过滤,滤饼用乙酸异丙酯淋洗。所得固体40℃鼓风干燥,得到类白色固体501.3g。收率90.9%。
[0084]
实施例5制备中间体int5(n=1)
[0085]
将495g int3和648.5g 2-甲基吡啶加至6l甲苯中,氮气置换。分3批加入292g五氯化磷,在35~45℃反应3h;反应后降温至0℃;滴加2n koh水溶液。滴毕,过滤,分液。
[0086]
有机相依次用纯化水洗涤,得有机相。
[0087]
向有机相中加入321.8g 1-(3-甲氧基苯基)哌嗪盐酸盐和3.3l水;搅拌下加入282.3g三乙胺。10~20℃反应1h。反应结束后过滤,分液;有机层用磷酸二氢钠水溶液洗涤,水洗涤;所得有机相在50℃减压浓缩,浓缩物加入51.83kg mtbe打浆,过滤,干燥,得509.8g类白色固体。收率73.3%。
[0088]
实施例6制备中间体int7(n=1)
[0089]
将300g int5和12.0g(r,r)-n,n'-双(三氟甲烷磺酰)-1,2-二苯基乙二胺加至1.5l丙酸甲酯中,55℃反应,反应结束后,将体系降温至25℃,搅拌下加入192g dtta,搅拌过夜。抽滤,用正庚烷淋洗,干燥。得到315.8g int7粗品。
[0090]
称取10.0g加至35ml的丙酸甲酯中,降至0~10℃,搅拌过夜。抽滤,干燥。得到类白色固体7.2g。收率:72%。
[0091]
实施例7制备终产品化合物i
[0092]
将4.5gint7加至17.5ml乙酸乙酯中,加入12.7g饱和碳酸氢钠水溶液,反应1h,分去水相。有机相浓缩。向浓缩物中加入丙酮和lioh水溶液(0.5glioh和20ml纯化水)。30℃反应4h。加至95ml纯化水中。用1m hcl调节ph至6.0。抽滤,湿品用纯化水淋洗烘干得产品2.7g,收率83.2%,纯度99.8%,ee值99.86%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1