一种视网膜分化潜能细胞系CRX-Shen001及其构建方法与流程
一种视网膜分化潜能细胞系crx-shen001及其构建方法
技术领域
1.本发明涉及基因工程领域,具体涉及一种具有人视网膜分化潜能的红色荧光标记细胞系及其构建方法。
背景技术:
2.视网膜退行性疾病包括遗传性视网膜变性、年龄相关性黄斑变性等。光感受器功能障碍和丧失是视网膜退行性疾病的主要原因之一。由于人类视网膜中的光感受器不可再生,视网膜退行性疾病会导致视杆和视锥细胞的永久性丧失。
3.目前临床上尚无针对视网膜退行性疾病的有效治疗方法。尤其是,视网膜退行性疾病的晚期阶段,在光感受器细胞已经大量丧失的情况下,目前的研究热点基因治疗和基因编辑方法均不适用。以恢复视网膜光敏性为目的的替代策略正处于探索阶段,包括光遗传工具、光感受器移植等。
4.光感受器移植是指将供体光感受器细胞移植到视网膜下腔从而补充替换已丧失的光感受器细胞,可以替代晚期视网膜变性中丢失的细胞,是一种很有前途的再生策略。影响其治疗效果的其中一个重要因素就是供体细胞的来源,确定合适的供体细胞类型和来源是至关重要的。
5.供体细胞由原代细胞或组织,发展到通过2d培养系统获得可移植的光感受器前体细胞,最终发展到今日通过3d培养技术获得视网膜类器官作为供体细胞来源。
6.光感受器的发育和维持需要精确调控的基因表达,这种调节是由以crx为中心的光感受器转录因子网络介导的,crx是一种同源结构域转录因子。crx是光感受器前体细胞的分子标志物,其可以作为筛选适合移植的光感受器前体细胞的途径,tdtomato是一种信号非常强的红色荧光蛋白,其对细胞和小鼠没有明显的毒性,是非常理想的细胞成像工具。因此,将tdtomato转入到干细胞染色体上的组织特异性的crx基因末端,可以得到能够在分化成视网膜类器官时表达红色荧光的干细胞。
7.crispr/cas是原核生物的一种免疫系统,最早于1987年被发现,随后,crispr/cas系统被运用成一种高效的基因编辑工具,成为第三代基因组定点编辑技术。2013年时,研究人员成功地使用crispr/cas系统编辑了哺乳动物细胞的基因组。由于其应用成本低,操作方便,效率高,一经出现便成为基因组定点编辑的热门技术。但是,crispr/cas系统介导的基因编辑得到的转基因细胞并非具有统一的性状,不同的实验批次,甚至同一批次的不同克隆,都有可能得到差异极大的结果。因此,需要在基因编辑后进行进一步的筛选,才能得到更符合研究需求的转基因细胞。
技术实现要素:
8.发明所要解决的技术问题是提供一种构建具有人视网膜分化潜能的红色荧光标记细胞系的方法,包括将红色荧光蛋白表达框插入到人干细胞基因组中的步骤。
9.在一个具体实施方案中,所述红色荧光蛋白为tdtomato。
10.在一个具体实施方案中,所述人干细胞为h9细胞。
11.在一个具体实施方案中,所述方法包括以下步骤:
12.s1:构建打靶载体,所述打靶载体包含所述红色荧光蛋白表达框和筛选用的抗性基因;
13.s2:通过crispr/cas9基因编辑系统将所述打靶载体插入到hes-zlm-001的第四外显子的终止密码子前;
14.s3:消除所述抗性基因。
15.在一个具体实施方案中,所述crispr/cas9基因编辑系统中sgrna的左侧向导序列为seq id no:1-3中的一个,右侧向导序列为seq id no:4-6中的一个。
16.在一个具体实施方案中,通过在所述打靶载体中搭载creert2系统来消除所述抗性基因。
17.本发明还提供了上述方法构建得到的具有人视网膜分化潜能的红色荧光标记细胞系。
18.本发明还提供了一种具有人视网膜分化潜能的红色荧光标记细胞,于2022年1月25日将crx-shen001保藏于中国湖北省武汉市武汉大学的中国典型培养物保藏中心(cctcc),保藏号cctcc no:c202207。
19.本发明通过设计打靶载体和相关的分子遗传操作,并通过筛选构建了一种红色荧光标记的胚胎干细胞系,该细胞系可经3d培养诱导分化为表达tdtomato红色荧光的视网膜类器官。得到的视网膜类器官同人类正常视网膜的神经细胞组成一致且发育的时间和空间顺序接近于正常的人类视网膜。该细胞系是一种强大的工具,可帮助实现人类视网膜发育和疾病产生的相关研究,并促进致盲疾病治疗方法的开发。
20.生物材料保藏
21.本发明的一个细胞系在2022年1月25日将crx-shen001保藏于中国湖北省武汉市武汉大学的中国典型培养物保藏中心(cctcc),保藏号cctcc no:c202207,命名为人源胚胎干细胞crx-shen001。
附图说明
22.图1为crispr/cas9介导crx-tdtomato-icreert2人胚胎干细胞系构建流程图。
23.图2为sgrna引物活性检测统计图。
24.图3为打靶载体质粒图。
25.图4为打靶载体酶切后的电泳照片。
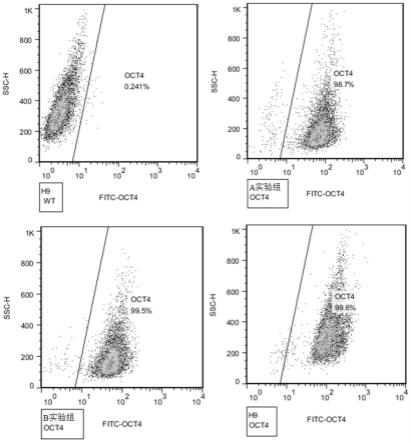
26.图5为各细胞系流式细胞分析,其中a组为a07细胞系的流式图,b组为crx-shen001细胞系的流式图。
27.图6为胚胎干细胞培养及诱导分化流程。
28.图7为a07细胞系(右)和crx-shen001细胞系(左)分化60天后得到的视网膜类器官的荧光显微照片。
具体实施方式
29.以下结合附图对发明的原理和特征进行描述,所举实例只用于解释发明,并非用
于限定发明的范围。
30.1、crispr/cas9介导红色荧光报告人胚胎干细胞系的构建
31.对h9细胞系靶位点序列进行pcr扩增并测序验证,确认其是否与genebank dna序列数据库和ensembl基因组数据库中所给序列一致。如图1所示,在hes-zlm-001基因exon4和3’utr之间终止密码子前插入p2a-tdtomato-p2a-icreert2,利用crispr/cas9技术制备hes-zlm-001敲进h9细胞系。
32.基于sgrna的设计原则,在靶位点区域共设计多条sgrna,并检测sgrna活性。
33.将293t细胞铺种96孔板,37℃,5% co2环境下培养,待密度为80%-90%时进行转染实验。通过lipofectamine 2000将pcs-sgrna和puca-msd共转染进293t细胞中。分设空白对照组:不加pcs-sgrna和puca-msd;阴性对照组:加puca-msd;阳性对照组:加阳性含有已知活性sgrna的pcs-sgrna和puca-msd;实验组:加含有需要进行活性检测sgrna的pcs-sgrna和puca-msd,各3个重复。37℃,5% co2环境下培养20-24小时。取上清,通过luc buffer i(10x),luc buffer ii(10x)和1mm腔肠素介导的萤火虫荧光信号显示体系处理,利用酶标仪检测各孔细胞上清液萤火虫荧光信号的强度。
34.结果如图2所示,5’端sgrna中,sgrna1、sgrna2、sgrna8活性较高。3’端sgrna中,sgrna11、sgrna12、sgrna13活性较高。使用上述三个sgrna进行下一步实验。
35.根据选择的sgrna构建打靶载体,经过酶切鉴定和测序,确认打靶载体构建完成。图3示出了打靶载体质粒图,图4示出了打靶载体酶切后的电泳图谱,结果显示,打靶载体酶切片段大小符合预期,说明打靶载体构建正确。
36.使用打靶载体对h9细胞系进行多批次的电转,得到大量转化子用于药物筛选、阳性克隆富集。设计引物同时扩增野生型和突变等位基因。挑单克隆进行pcr测序,根据测序结果和测序峰图判断具体基因型:纯合/杂合/野生型,得到未去抗性敲进阳性克隆。
37.将未去抗性阳性克隆消化为单细胞,使用电转缓冲液悬浮单细胞,使其浓度为1x 104个/μl。取100μl单细胞悬液,加入10μg pcag-icre载体,于1100v,40ms,1pulse的参数下进行电转。电转后的细胞于37℃,5%co2环境下培养。48小时后进行更昔洛韦药物筛选。所得克隆再次进行pcr测序,最终得到去抗性敲进阳性克隆,得到了一些去抗性敲除阳性克隆,综合考虑pcr结果及细胞株状态后,最终筛选出多个去抗性敲进阳性克隆。
38.2、流式细胞分析验证细胞系干性
39.分别向h9细胞系及上述转基因细胞系的培养皿内加入edta消化液,孵育6-8分钟,1200rpm离心5分钟,去上清,用dpbs培养基重悬细胞,细胞计数。根据细胞计数结果计算取液量,将细胞悬液转移至离心管内,保证每个离心管内细胞数为1~5x107个,h9细胞悬液转移至两个离心管中,转基因克隆细胞悬液分别转移至一个离心管中。将离心管内的细胞悬液1200rpm离心5分钟,去上清,用质量分数4% pfa在4℃固定1小时,1200rpm离心5分钟去上清,pbs清洗后1200rpm离心5分钟去上清;用体积分数4% bsa和2% tri在4℃封闭过夜,1200rpm离心5分钟去上清;一管h9细胞悬液和转基因克隆细胞悬液内加入oct4一抗,4℃孵育24小时,1200rpm离心5分钟去上清,pbs清洗后1200rpm离心5分钟去上清;向离心管内加相应荧光标记二抗,4℃避光孵育过夜,1200rpm离心5分钟去上清,pbs清洗后1200rpm离心5分钟去上清。用pbs重悬细胞,200目尼龙网过滤后将其转移至流式管中,上机检测。
40.两管h9细胞分别作为阳性对照组和阴性对照组,转基因克隆细胞作为实验组,观
察其oct4阳性细胞比例。
41.结果如图5所示,阴性对照组的oct4
+
细胞占0.241%,阳性对照组为99.6%。1-a07(本团队已报道)和crx-shen001实验组中,oct4
+
细胞比率分别为98.7%和99.5%,说明这两个实验组的细胞系干性均良好。其中,以crx-shen001的细胞干性比1-a07更好。
42.进行细胞爬片实验和核型分析,结果显示,1-a07和crx-shen001可成功表达干细胞标志物sox2、nanog、ssea4,染色体数量和形态无异常,表明细胞系干性良好,可用于增殖、分化等培养。
43.3、人胚胎干细胞经3d培养诱导分化为视网膜类器官
44.将上述细胞系进行3d培养诱导分化为视网膜类器官,诱导流程如下如图6所示。
45.取6ml d-mem/f12与100μl基质胶于15ml离心管中,充分混匀,取1ml加入培养皿。把培养皿放到培养箱里,静置30分钟,吸去液体,加入1ml mtesr1培养基和2μl y-27632hcl。待水浴锅升温至37℃,从液氮罐中取出冻存的胚胎干细胞,将冻存管在水浴锅中晃动速溶。取5ml mtesr1于15ml离心管,加入干细胞液混匀。200g离心5分钟,弃上清。取1ml mtesr1重悬细胞后转移至培养皿,摇匀。
46.镜下观察细胞的状态、密度后,放入培养箱。之后每隔24小时取出培养皿,在镜下观察细胞状态、密度后,吸去培养基,加入1ml dpbs,摇晃后吸去液体,洗去其中悬浮的死细胞。加入2ml mtesr1培养基后放回培养箱。待人胚胎干细胞融合度达到70%-80%时进行诱导分化。诱导分化人胚胎干细胞,将人胚胎干细胞酶解成单细胞,用含y-27632的medium i培养基重悬细胞,置于3d细胞培养96孔板中,细胞接种密度为9000个/孔,每孔100μl。当天记录为分化第0天;分化第2天向每孔加入质量分数1%基质胶;分化第6天用不含y-27632的medium i培养基更换孔内一半原培养基;分化第12天将每孔内的拟胚体转移到细菌培养皿中,使用含1%基质胶的medium ii培养基继续培养;分化第18天将拟胚体切割成4~5小块后,转移到新的细菌培养皿中,使用含视黄酸的medium iii培养基继续培养,之后每7天换液1次。medium i、ii、iii培养基分别对应图中分化第0天至第12天、分化第12天至第18天、分化第18天以后所用培养基。
47.上述诱导实验进行了5个批次,每组每个批次对50个细胞进行诱导,结果如表1所示,培养到60天时,a07细胞系平均每批次诱导的得到29个视网膜类器官,而crx-shen001平均每批次诱导得到38个视网膜类器官。可见,crx-shen001可诱导分化成视网膜类器官的成功率高31%。
48.表1诱导实验获得的视网膜类器官数量统计
49.批次12345平均crx-shen001452839423538a07302521274029
50.对分化第60天的视网膜类器官进行荧光显微镜观察室,结果如图7所示,a07分化的类器官冰冻切片tdtomato荧光表达主要分布于中深层,少量分布于顶层,而crx-shen001具有明显更为强烈的荧光,并且主要分布于顶层。上述结果表明,我们制备的crx-shen001细胞系经诱导分化得到的视网膜类器官可成功表达tdtomato红色荧光,且红色荧光大部分分布于视网膜类器官顶层,符合正常人视网膜中光感受器前体细胞分布规律,可用于指示该经诱导分化得到的视网膜类器官中的光感受器前体细胞。
51.基于以上实验,我们发现,crx-shen001具有明显更好的细胞干性,以及更强烈的tdtomato荧光表达,并且荧光表达符合正常人视网膜中光感受器前体细胞分布规律。因此,我们认为,crx-shen001可更好地用于人类视网膜发育和疾病产生的相关研究,并促进致盲疾病治疗方法的开发。因此,我们在2022年1月25日将crx-shen001保藏于中国湖北省武汉市武汉大学的中国典型培养物保藏中心(cctcc),保藏号cctcc no:c202207,命名为人源胚胎干细胞crx-shen001。
52.尽管上面已经示出和描述了发明的实施例,可以理解的是,上述实施例是示例性的,不能理解为对发明的限制,本领域的普通技术人员在发明的范围内可以对上述实施例进行变化、修改、替换和变型。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1