一种提高基因敲入效率的电穿孔转染体系制备方法与流程
1.本技术涉及生物技术领域,特别涉及一种提高基因敲入效率的电穿孔转染体系制备方法。
背景技术:
2.基因敲入(gene knock in)是利用基因同源重组,将外源的功能基因(基因组原先不存在、或已失活的基因),转入细胞与基因组中的同源序列进行同源重组,插入到基因组中,在细胞内获得表达的技术。该技术在基因功能与分子机制的研究、构建人类疾病的动物模型、基因治疗、农业和林业等领域具有重要的应用前景。以talen、crispr/cas系统为代表的人工核酸内切酶技术在制备indel(插入或缺失标记)等基因突变体的应用中已较为成熟,但在基因敲入的应用中仍面临着编辑效率低的困境。基因编辑系统的递送策略是影响基因敲入(ki)效率的关键因素之一,为了获得最佳的ki效率,已发展了不同的转染方案,其中,电穿孔转染方法是一种高效的物理方法,可以实现对基因编辑系统高效的细胞内或细胞核内递送,使用细胞范围广,特别是针对难转染的干细胞、神经细胞等。
3.电穿孔转染细胞的方法是利用瞬间高压造成细胞膜不稳定,形成电穿孔使得外源dna、rna或蛋白等进入细胞。电穿孔转染法是一种物理方法,无生物与化学毒性副作用,安全性高,操作方便,但其转染效率也受多种因素影响,除常规的电压、脉冲时间、电穿次数等因素外,很大程度上还取决于待转染组分的溶液成分。通常情况下,电转仪配有专用的电转液,待转染的组分、细胞和电转液需严格按照特定的比例添加以制备具有最佳转染效率的转染体系,因此对质粒dna、mrna或蛋白等待转染物质的制备浓度要求较高,只有待转染物质的浓度极高时,才能在满足转染体系制备比例的情况下,具有足够的量获得较高的编辑效果。同时,在用移液器吸取高浓度待转物质时,由于微量的体积和移液吸头的吸附作用,会导致组分实际用量误差,从而导致转染效率不稳定,影响实验结果。这种转染体系的制备方法要求高、操作也较为繁琐,不利于使用。
技术实现要素:
4.鉴于以上所述现有技术的缺点,为解决现有技术中电转染效率低,基因敲入不稳定的技术问题,本技术的目的在于提供一种提高基因敲入效率的电穿孔转染体系制备方法,用于解决现有技术中的问题。
5.为实现上述目的及其他相关目的,本技术第一方面提供一种电转染方法,包括以下步骤:
6.1)提供靶向目标基因的crispr/cas9系统;
7.2)提供靶向目标基因的同源修复模板;
8.3)将步骤1)所得的crisrp/cas9系统和步骤2)所得的同源修复模板干燥成粉末,得到预处理样品;
9.4)将步骤3)中的预处理样品用电转缓冲溶液重悬,与待转入的细胞共孵育,形成
电转染体系;
10.5)将步骤4)所得的电转染体系进行电击处理。
11.本技术第二方面提供前述的电转染方法用于体外基因敲入的用途。
12.与现有技术相比,本技术的有益效果为:
13.1、本技术可显著提高基因敲入的效率,适用于不同基因的敲入。
14.2、本技术可最大程度地减少电转体系组分的取样误差,降低实验误差,增加实验结果的稳定性。
15.3、本技术对crispr/cas系统的制备浓度要求低,不限crispr/cas系统的任何形式。
16.4、本技术适用于任何电转仪器设备的电转体系,不限电转设备品牌和型号。
附图说明
17.图1显示为px330质粒图谱。
18.图2显示为dsdna hdr 1%琼脂糖凝胶电泳图。其中,m1表示1000bp dna marker,m2表示100bp dna marker,泳道1和泳道2均为对照组dna,泳道3为dsdna hdr。
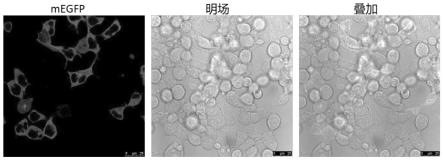
19.图3显示为实验组实施例基因敲入效果。
20.图4显示为对比例1基因敲入效果。
21.图5显示为对比例2基因敲入效果。
具体实施方式
22.为了使本技术的发明目的、技术方案和有益效果更加清晰,下面结合实施例对本技术作进一步说明。应理解,所述实施例只用于解释本技术,并非用于限定申请的范围。下述实施例中所使用的试验方法如无特殊说明,均为常规方法,熟悉此技术的人士可由本说明所揭露的内容容易地了解本技术的其他优点及功效。
23.本技术所公开的“范围”以下限和上限的形式来限定,给定范围是通过选定一个下限和一个上限进行限定的,选定的下限和上限限定了特别范围的边界。这种方式进行限定的范围可以是包括端值或不包括端值的,并且可以进行任意地组合,即任何下限可以与任何上限组合形成一个范围。例如,如果针对特定参数列出了60~120和80~110的范围,理解为60~110和80~120的范围也是预料到的。此外,如果列出的最小范围值1和2,和如果列出了最大范围值3,4和5,则下面的范围可全部预料到:1~3、1~4、1~5、2~3、2~4和2~5。在本技术中,除非有其他说明,数值范围“a~b”表示a到b之间的任意实数组合的缩略表示,其中a和b都是实数。例如数值范围“0~5”表示本文中已经全部列出了“0~5”之间的全部实数,“0~5”只是这些数值组合的缩略表示。另外,当表述某个参数为≥2的整数,则相当于公开了该参数为例如整数2、3、4、5、6、7、8、9、10、11、12等。
24.如果没有特别的说明,本技术的所有步骤可以顺序进行,也可以随机进行,优选是顺序进行的。例如,所述方法包括步骤1)和2),表示所述方法可包括顺序进行的步骤1)和2),也可以包括顺序进行的步骤2)和1)。
25.本技术的发明人经过大量探索研究,发现了一种提高基因敲入效率的电穿孔转染体系制备方法,显著提高基因敲入效率。在此基础上完成了本技术。
26.本技术提供一方面提供一种电转染方法,包括以下步骤:
27.1)提供靶向目标基因的crispr/cas9系统;
28.2)提供靶向目标基因的同源修复模板;
29.3)将步骤1)所得的crisrp/cas9系统和步骤2)所得的同源修复模板干燥成粉末,得到预处理样品;
30.4)将步骤3)中的预处理样品用电转缓冲溶液重悬,与待转入的细胞共孵育,形成电转染体系;
31.5)将步骤4)所得的电转染体系进行电击处理。
32.本技术所提供的电转染方法中,步骤1)中,crispr/cas9系统包括cas9和sgrna。进一步地,crispr/cas9系统包括分别表达cas9和sgrna的质粒、cas9 mrna和sgrna,或cas9 rnp蛋白复合物。更进一步地,表达cas9的质粒选自pmj920和pcag-t3-hcas-pa。表达sgrna的质粒选自sp6-sgrna-scaffold。cas9 mrna和sgrna的mrna选自px330、px458和px459质粒中的rna。本发明对于crispr/cas9系统没有特殊要求,使用质粒、mrna或蛋白等任何形式均能实现高效的基因敲入效果。
33.crispr-cas9系统就是通过人工设计的sgrna(guide rna)来识别目的基因组序列,并引导cas9蛋白酶进行有效切割dna双链,形成双链断裂,损伤后修复会造成基因敲除或敲入等,最终达到对基因组dna进行修饰的目的。
34.本技术所提供的电转染方法中,步骤1)中,当crispr/cas9系统包括分别表达cas9的质粒和sgrna的质粒时,表达cas9的质粒和表达sgrna的质粒的质量比为1:0.5~4:1;优选为2:1。以1个24孔板的反应体系为基准,表达cas9的质粒和表达sgrna的质粒的浓度范围均为10~2000ng/μl;优选地,为10~100ng/μl、100~1000ng/μl、或1000~2000ng/μl等。本技术对crispr/cas系统的制备浓度要求低。
35.本技术所提供的电转染方法中,步骤1)中,当crispr/cas9系统为cas9 mrna和sgrna时,cas9 mrna和sgrna的质量比为2:1~6:1;优选为2:1。以1个24孔板的反应体系为基准,cas9 mrna和sgrna的制备浓度范围均为10~2000ng/μl;优选地,为10~100ng/μl、100~1000ng/μl、或1000~2000ng/μl等。本技术对crispr/cas系统的制备浓度要求低。
36.本技术所提供的电转染方法中,步骤1)中,当crispr/cas9系统为cas9 rnp蛋白复合物时,cas9蛋白和sgrna的摩尔量的比为1:1~1:1.5;优选为1:1.2。以1个24孔板的反应体系为基准,cas9蛋白和sgrna的制备浓度范围均为100μg/ml~10mg/ml;优选地,为100μg/ml~1mg/ml、1mg/ml~5mg/ml或5mg/ml~10mg/ml等。本技术对crispr/cas系统的制备浓度要求低。
37.本技术所提供的电转染方法中,步骤2)中,同源修复模板选自质粒模板、双链dna模板、线性单链dna模板和环形单链dna模板。换句话说,本发明对于同源修复模板的形式没有特殊要求,根据具体实验需求选择任何形式的模板,均能实现高效的基因敲入效果。
38.同源修复模板是由需要导入的目标基因和靶序列上下游的同源性序列(同源臂)组成,同源臂的长度和位置由编辑序列的大小决定。同源修复模板可以通过同源重组将目标基因插入靶向位点,从而将一段dna序列精准地插入特定的位点。
39.当使用质粒dna模板时,不限制质粒dna载体的选择,可包括含任何筛选基因的质粒dna载体。当使用双链dna模板时,不限制双链dna模板的制备方法,包括pcr扩增法,化学
合成法等。当使用线性单链dna模板时,不限制线性单链dna制备方法,包括化学合成法、酶降解法、磁珠法、pecan法等;优选地,为pecan法。
40.本技术所提供的电转染方法中,步骤1)所提供的crispr/cas9系统和步骤2)所提供的同源修复模板例如可以溶解在ddh2o中。
41.用crispr/cas9系统进行基因敲入的过程中,当dna双链断裂后,如果有dna修复模板进入到细胞中,基因组断裂部分会依据修复模板进行同源重组修复(hdr),从而实现基因敲入。在该修复路径中,需要将一段与靶标位点上下游紧邻序列的同源臂、特异的grna和cas9核酸酶一起引入细胞中。
42.本技术所提供的电转染方法中,步骤3)中,干燥选自冷冻真空干燥和/或旋转蒸发干燥。当选择冷冻真空干燥时,温度为-10~-80℃,优选地,为-80℃。当选择旋转蒸发干燥时,温度为4~37℃,旋转转速为50~160转/分;优选地,温度为4℃。预处理样品后,将携带有目标基因的crisrp/cas9系统和与之配对的同源修复模板实现体积最小化,便于控制用电转缓冲液重悬的体积,同时减少电转过程中吸取样品带来的误差,从而实现高效的基因敲入,并提高电转染的稳定性。
43.本技术所提供的电转染方法中,步骤4)中,待转入的细胞例如可以是悬浮、半悬浮和贴壁的动物细胞,还可以是易转染和难转染的动物细胞。细胞可以是本领域已知的各种细胞,包括但不限于免疫细胞、干细胞、细胞株等。所述细胞优选是哺乳动物来源的细胞,更优选为人细胞。一些实施方式中,所述细胞选自外周血细胞、造血细胞、神经干细胞和肿瘤细胞。一些实施方式中,所述细胞选自胚胎肾细胞、红细胞、中性粒细胞、嗜酸性粒细胞、嗜碱性粒细胞、外周血单核细胞或淋巴细胞等。
44.本技术所提供的电转染方法中,步骤4)中,孵育的温度为25~37℃,孵育的时间为5~30min。优选地,孵育温度为37℃,孵育时间为15min。
45.本技术所提供的电转染方法中,和/或,步骤4)中,电转缓冲液选自thermo fisher neon的buffer r、buffer t、lonza nuclefector的p1、p2、p3、p4、p5、se、sf、sg和biorad gene pulser xcell的gene pulser electroporation buffer中的一种或多种的组合。本技术所用的电转缓冲液与电转设备配套使用,例如当电转设备为thermo fisher neon时,所用电转缓冲液为buffer r或buffer t;当电转设备为lonza nuclefector时,电转缓冲液为适用于原代细胞的p1、p2、p3、p4、p5和适用于细胞系的se、sf和sg;当电转设备为biorad gene pulser xcell时,电转缓冲液为gene pulser electroporation buffer(货号1652677和1652676)。
46.本技术所用的电转缓冲液也可以是本领域技术人员熟知的其他电转缓冲液,例如可以为细胞培养液、磷酸缓冲液和cytomix缓冲液。优选地,细胞培养液选自rpmi1640、dmem、dmem/f12、或opti-mem;磷酸缓冲液选自k-pbs,hbs,hebs或pbs;cytomix缓冲液包括kcl、cacl2、k2hpo4、hepes、egta、mgcl2、atp和谷胱苷肽中的一种或多种。
47.本技术所提供的电转染方法中,步骤5)中,电转染仪器设备选自市售或自主开发的所有电转染系统。换句话说,本发明对于电转染系统没有特殊要求,如可选择thermo fisher的neon转染系统、lonza nuclefector系列,以及biorad的gene pulser xcell真核电穿孔系统等,均可以实现高效的基因敲入效率。
48.本技术另一方面提供利用所述的电转染方法用于体外基因敲入的用途。
49.下面通过实施例对本技术予以进一步说明,但并不因此而限制本技术的范围。
50.具体实施例以靶向hek293t细胞内tuba1b基因敲入megfp荧光标签蛋白为例,进一步阐述说明本发明专利的制备方法。crispr/cas系统选择cas9蛋白和sgrna共表达质粒px330质粒系统,同源修复模板选用dsdna。电转设备选择thermo fisher的neon转染系统。
51.实施例1靶向tuba1b基因的crispr/cas系统的制备
52.1、sgrna双链片段的合成。靶向人tuba1b基因的sgrna序列(选自参考文献:theodore l.roth.et al.,reprogramming human t cell function and specificity with non-viral genome targeting,nature,559:405
–
4092018)为:5
’‑
tggagatgcactcacgctgc-3’,根据sgrna靶向的序列,合成一对序列互补的ssdna(生工生物工程(上海)股份有限公司合成),其序列如下:
53.sg-r:5
’‑
caccgtggagatgcactcacgctgc-3’(seq id no:1)
54.sg-f:5
’‑
aaacgcagcgtgagtgcatctccac-3’(seq id no:2)
55.将两条ssdna退火成双链dna(dsdna)。
56.退火体系(20μl)如下:sg-r(100μm)1μl;sg-f(100μm)1μl;灭菌水18μl。退火程序如下:1)95℃保持5min;2)95℃到25℃,以-1℃/s的速率降低温度,共执行70个循环;3)25℃保持7min;4)4℃保存。退火结束后,待用。
57.2、线性化px330质粒载体(addgene plasmid#42230,图谱见图1)。px330为cas9蛋白和sgrna共表达的载体质粒,使用bbsi酶进行单酶切以获得线性化的质粒载体。
58.3、连接退火dsdna和线性化质粒。使用t4 dna连接酶将步骤1和2中的退火dsdna片段和线性化px330质粒按t4 dna连接酶试剂盒(neb,货号#m0202l)方法进行连接。将连接产物转化到大肠杆菌dh5a感受态细胞中,涂布于含氨苄抗性的lb平板中,过夜培养后,挑取单克隆。将单克隆进行扩大培养,并使用质粒提取试剂盒(天根生化科技有限公司,货号dp103)提取单克隆重组质粒,然后测序以鉴定阳性单克隆。同时,使用超微量紫外分光光度计进行定量。
59.结果:构建好的重组质粒测序结果如seq id no:3所示,其中下划线和加粗标记的为tuba1b基因的sgrna靶向序列,表明成功构建了靶向tuba1b基因的crispr/cas9系统表达质粒。重组质粒定量结果为800ng/μl。
60.靶向hek293t细胞tuba1b基因的px330-tuba1b重组质粒序结果:
61.62.63.64.[0065][0066]
实施例2megfp荧光标签的dsdna同源修复模板制备
[0067]
使用pcr扩增的方法制备获得靶向tuba1b基因敲入megfp荧光标签的dsdna同源修复模板,其中aicsdp-4:tuba1b-megfp质粒(addgene plasmid#87421)为pcr扩增体系的模板,根据上下游同源臂的序列设计上下游引物,其中,同源修复模板长度为1570bp,其序列信息见seq id no:4:
[0068]5’‑
aggaggccttcccggtttaggatgggaaggtaacattcattaaaagcaacgtagact atagtgtagctgttctcaaaagtagtacatcttagaaaaggatctttagaaaagatcgctttagaaaaggaaattcgttttcagattacgtgagtagcctaggtaacacagccagacctcatctccacaaaaaaaatgaaaaaattagccagcttggtggtctgtgcctgtggtcccagctgctccagaggctgaggtggggggatgactggagcctaggctgcagtgagcctagatggcatcactgcactcaagcctgggcgacagaccttatctctaaaaaaataaagattgcatgagtattttgttcc
acttgacagtcatcaatagattggtttaaattgtgatatcttttttacttaccgcaggtgagcaagggcgaggagctgttcaccggggtggtgcccatcctggtcgagctggacggcgacgtaaacggccacaagttcagcgtgtccggcgagggcgagggcgatgccacctacggcaagctgaccctgaagttcatctgcaccaccggcaagctgcccgtgccctggcccaccctcgtgaccaccctgacctacggcgtgcagtgcttcagccgctaccccgaccacatgaagcagcacgacttcttcaagtccgccatgcccgaaggctacgtccaggagcgcaccatcttcttcaaggacgacggcaactacaagacccgcgccgaggtgaagttcgagggcgacaccctggtgaaccgcatcgagctgaagggcatcgacttcaaggaggacggcaacatcctggggcacaagctggagtacaactacaacagccacaacgtctatatcatggccgacaagcagaagaacggcatcaaggtgaacttcaagatccgccacaacatcgaggacggcagcgtgcagctcgccgaccactaccagcagaacacccccatcggcgacggccccgtgctgctgcccgacaaccactacctgagcacccagtccaagctgagcaaagaccccaacgagaagcgcgatcacatggtcctgctggagttcgtgaccgccgccgggatcactctcggcatggacgagctgtacaagggaggttcaggaggcagcgagtgcatctccatccacgttggccaggctggtgtccagattggcaatgcctgctgggagctctactgcctggaacacggcatccagcccgatggccagatgccaagtgacaagaccattgggggaggagatgactccttcaacaccttcttcagtgagacgggcgctggcaagcacgtgccccgggctgtgtttgtagacttggaacccacagtcattggtgagttgacctcagtaacctgagatcccaggatgctgggacaggaggtctgtccaggggcttctcttgtcactcactcactccctccgtccttctctccctcctccagatgaagttcgcactggcacctaccgccagctcttccaccctgagcagctcatcacaggcaaggaagatgctgccaataactatg-3’(seq id no:4)。
[0069]
上下游引物序列如下:
[0070]
上游引物5
’‑
agaggccttcccgg-3’(seq id no:5)
[0071]
下游引物5
’‑
catagttattggcagcatcttccttg-3’(seq id no:6)
[0072]
pcr反应使用phanta super-fidelity dna聚合酶(南京诺唯赞生物科技股份有限公司,货号p501-d1),pcr反应体系为:
[0073][0074][0075]
pcr反应程序为:
[0076][0077]
经pcr反应后,扩增出dsdna同源修复模板(dsdna hdr)。pcr反应产物用pcr产物抽提试剂盒(takara,9761)进行纯化,获得的dsdna hdr用ddh2o溶解,使用超微量紫外分光光
度计进行定量后,保存于-20℃,待用。同时,使用1%琼脂糖凝胶电泳验证dsdna hdr的制备质量。
[0078]
结果:制备好的dsdna同源修复模板定量结果为定量结果为500ng/μl,1%琼脂糖凝胶电泳结果见图2,表明成功制备了长度为1570bp,高纯度的靶向tuba1b基因敲入megfp荧光标签的dsdna同源修复模板。
[0079]
实施例3靶向tuba1b基因敲入megfp荧光蛋白标签的电转染体系制备
[0080]
细胞培养:人胚胎肾细胞(hek293t)购自中国科学院典型培养物保藏委员会(上海)细胞库(目录号为scsp-502)。培养条件为含有10% fbs(灭活,56℃/30min)、50units/ml青霉素、50μg/ml链霉素和4mm谷氨酰胺的dmem培养基(gibco),置于恒温二氧化碳培养箱(37℃,5% co2)中培养的。其传代比例为1:4,离心条件为1000rpm,3min。
[0081]
按24孔细胞培养板的培养条件,设计靶向tuba1b基因敲入megfp荧光标签的电转染体系,主要组分如下:px330重组质粒500ng,dsdna hdr 2500ng,hek293t细胞18
×
104个重悬在12μl buffer r(thermo fisher,货号mpk1096)中。制备方法如下:
[0082]
1、分别量取0.625μl浓度为800ng/μl实施例1获得的px330重组质粒(总量为500ng)和5μl 500ng/μl实施例2获得的dsdna hdr(总量为2500ng)置于1.5ml离心管中,使用冷冻浓缩离心干燥器(华美生化仪器,型号lng-t98)在4℃,1000rpm的条件下干燥30min,获得px330重组质粒和dsdna hdr的混合物粉末。然后,使用10μl buffer r重悬该混合物粉末,并置于37℃恒温培养箱中孵育15min。
[0083]
2、从培养皿中消化hek293t细胞,用培养基洗脱hek293t细胞,1000rpm离心3min后,弃培养基,用pbs缓冲溶液重悬细胞,细胞计数后,取18
×
104个细胞的量置于新的1.5ml离心管中,1000rpm离心3min后,弃pbs缓冲溶液,使用步骤1中获得10μl buffer r重悬混合物粉末重悬细胞,并补加buffer r溶液,使得体系的总体积为12μl。
[0084]
结果:按照以上实验过程,即可制备获得靶向tuba1b基因敲入megfp荧光标签的电转染体系。
[0085]
实施例4靶向tuba1b基因敲入megfp荧光标签的电转染效果
[0086]
电转实验例:
[0087]
电转体系同实施例3。将电转体系根据neon电转系统的操作步骤(参考文献,xiquan liang,et al.,enhanced crispr/cas9-mediated precise genome editing by improved design and delivery of grna,cas9 nuclease,and donor dna.journal of biotechnology.2017,241:136-146.),在1100ms的脉冲宽度、30v的电压、1次脉冲的电转条件下进行电击,将电击后的细胞转移至已含有500μl培养基的24孔板中,37℃继续培养48h后,通过流式细胞仪统计基因敲入效率,在共聚焦显微镜下观察敲入效果。基因敲入效率(%)=gfp细胞/总细胞*100%。该实验重复3次。
[0088]
电转对比例1:
[0089]
1、取0.625μl浓度为800ng/μl实施例1获得的px330重组质粒(总量为500ng)和5μl浓度为500ng/μl实施例2获得的dsdna hdr(总量为2500ng)置于1.5ml离心管中,5.625μl混合物37℃恒温培养箱中孵育15min。
[0090]
2、细胞培养同实施例3。培养皿中消化hek293t细胞,用培养基洗脱hek293t细胞,1000rpm离心3min后,弃培养基,用pbs缓冲溶液重悬细胞,细胞计数后,取18
×
104个细胞的
量置于新的1.5ml离心管中,1000rpm离心3min后,弃pbs缓冲溶液,使用步骤1中获得5.625μl混合物溶液,并补加buffer r溶液,使得电转体系的总体积为12μl。
[0091]
3、将电转体系根据neon电转系统的操作步骤,参数与电转实验例相同。
[0092]
电转对比例2:
[0093]
neon电转系统中对一个24孔板电转体系的要求为:总电转体系12μl,其中电转buffer r缓冲液的体积至少为10μl,核酸和细胞等额外的组分不超过2μl,才能达到理想的转染效果。为满足此电转体系的要求,需对实施例1获得的px330重组质粒和实施例2获得的dsdna hdr进行浓缩至高浓度,使得这些组分的体积足够少。
[0094]
1、浓缩转染组分:分别将实施例1获得的px330重组质粒和实施例2获得的dsdna hdr置于冷冻浓缩离心干燥器(华美生化仪器,型号lng-t98)4℃,1000rpm的条件下干燥10min中浓缩至1000ng/μl和5000ng/μl。
[0095]
2、取0.5μl浓度为1000ng/μl步骤1获得的px330重组质粒(总量为500ng)和0.5μl浓度为5000ng/μl步骤1获得的dsdna hdr(总量为2500ng)置于1.5ml离心管中,1.0μl混合物37℃恒温培养箱中孵育15min。
[0096]
3、细胞培养同实施例3。培养皿中消化hek293t细胞,用培养基洗脱hek293t细胞,1000rpm离心3min后,弃培养基,用pbs缓冲溶液重悬细胞,细胞计数后,取18
×
104个细胞的量置于新的1.5ml离心管中,1000rpm离心3min后,弃pbs缓冲溶液,使用步骤2中获得1.0μl混合物溶液,并补加buffer r溶液,使得电转体系的总体积为12μl。
[0097]
4、将电转体系根据neon电转系统的操作步骤,参数与电转实验例相同。
[0098]
结果:基因敲入效率统计结果见表1,其中实验例敲入效率最高,为20.43%,对比例1没有实现基因敲入,对比例2敲入效率为8.57%。同时,实验例敲入效率稳定,3次重复实验的基因敲入效率方差为0.12%,而对比例3次重复实验之间敲入效率波动大,不稳定。
[0099]
表1基因敲入效率统计结果
[0100][0101]
基因敲入的共聚焦显微成像结果如图3、图4和图5所示,实验例阳性敲入hek293t细胞最多,对比例2有较少的阳性敲入细胞,对比例3没有观察到阳性敲入细胞。
[0102]
以上实验结果表明,本发明中的基因敲入电穿孔转染体系制备方法可实现稳定且高效的基因敲入。
[0103]
上述实施例仅例示性说明本发明的原理及其功效,而非用于限制本技术。任何熟悉此技术的人士皆可在不违背本技术的精神及范畴下,对上述实施例进行修饰或改变。因此,举凡所属技术领域中具有通常知识者在未脱离本发明所揭示的精神与技术思想下所完成的一切等效修饰或改变,仍应由本技术的权利要求所涵盖。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1