一种3-N-乙酰依替米星的制备方法与流程
一种3-n-乙酰依替米星的制备方法
技术领域:
1.本发明涉及药物化学领域,一种氨基糖苷类化合物的制备方法,具体来说是一种3-n-乙基庆大霉素c1a的制备方法。
背景技术:
2.硫酸依替米星(etimicin sulfate)为半合成氨基糖苷类抗生素,适用于对其敏感的大肠杆菌、肠杆菌属、克雷伯氏肺炎杆菌、沙雷氏杆菌属、不动杆菌属、枸橼酸杆菌、变形杆菌属、绿脓杆菌、嗜血流感杆菌和葡萄球菌等引起的各种感染。临床研究显示硫酸依替米星制剂对以下感染有较好的疗效,如呼吸道感染,肾脏和泌尿生殖系统感染,皮肤软组织和其它感染等。
3.3-n-乙酰依替米星,结构式如下:
[0004][0005]
3-n-乙酰基依替米星是硫酸依替米星原料药生产过程中的杂质之一,其存在不仅会影响到依替米星收率,对硫酸依替米星原料药质量也存在潜在风险。由于与依替米星结构相似,性质相近,较难从生产中分离。目前国内外均没有3-n-乙酰基依替米星的标准品出售。
[0006]
目前有关于3-n-乙酰基依替米星制备方法的专利(中国专利申请号:202011536901.4),其主要步骤为:在圆底烧瓶中投入依替米星、乙醇、水搅拌均匀,降温至0℃,滴加二碳酸二叔丁酯,持续搅拌,反应完成后,将反应体系浓缩;反应体系中加入甲酸,升温至120℃,持续反应4h,反应结束后降温至室温;分别加入10%氢氧化钠,搅拌加热回流,反应结束后,降温至室温;除盐,通过分离的到目标产物3-n-乙酰基依替米星。该制备方法boc保护基不具有选择性,无法区分2
′‑
c-氨基和6
′‑
c-氨基和3-c-氨基,致使制备过程中必定产生副反应,收率低,且在用10%的氢氧化钠溶液脱保护时,由于反应时间过久,大量的3-n-乙酰基也会被破坏掉,造成收率进一步降低,同时因为同分异构体存在,分离难度更大,导致产率低或者纯度低等问题。
技术实现要素:
[0007]
本发明的目的在于提供一种3-n-乙酰基依替米星的制备方法。该方法具有专属性好副反应少,易于纯化分离的优点。
[0008]
本发明所述的制备方法,包括以下步骤:
[0009]
步骤1)庆大霉素c1a和乙酸锌反应得锌络合物;
[0010]
步骤2)三乙胺作为缚酸剂,加入乙酸酐反应后得到氨基被乙酰化的产物;
[0011]
步骤3)与草酸钠反应解开络合,经分离纯化得p1中间体;
[0012]
步骤4)加入乙二醇二甲醚、六甲基二硅氮烷和浓硫酸反应,得羟基硅烷化产物;步骤5)浓缩除溶剂后再加入二氯甲烷和乙醛反应,随后加入硼氢化钾和硼酸缓冲液反应得到乙基化物;
[0013]
步骤6)用氢氧化钠溶液水解,水解液过柱,制备液相分离纯化得到3-n-乙酰基依替米星。
[0014]
所述方法,制备路线如下:
[0015][0016]
本发明方法,核心在于步骤6)的水解,水解液过柱,纯化步骤。
[0017]
其中,所述水解步骤如下:
[0018]
反应液6中加入10%氢氧化钠溶液100ml,浓缩蒸去二氯甲烷,再加入20%氢氧化钠240ml溶液,升温至110℃回流,搅拌3小时,得反应液7。
[0019]
其中,所述水解液过柱步骤如下:
[0020]
反应液7以大孔树脂吸附,以无盐水冲洗除盐,以5%~25%乙醇梯度洗脱,收集纯度>80%的洗脱液,真空浓缩后得到粗品。
[0021]
其中,所述纯化,采用制备液相分离纯化,步骤如下:
[0022]
取2g粗品置于离心管中,用流动相a溶解,按照以下制备液相条件进行:
[0023][0024]
得3-n-乙酰基依替米星纯品,经过检测,3n-乙酰依替米星面积归一化含量为97.18%(elsd)。
[0025]
本发明的方法,相对于现有的工艺而言,具有以下有益效果:
[0026]
通过本发明的方法制备得到的3-n-乙酰基依替米星有专属性好副反应少,产品易于分离纯化,产率高等特点。同时,对该杂质进行深入研究,通过优化水解工艺,降低该杂质生成,对于提高药品质量,提高临床用药的安全性具有重大的意义。
[0027]
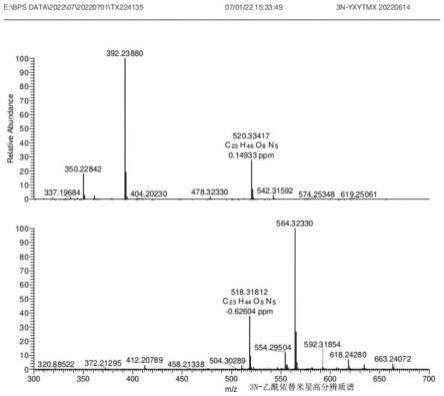
以下通过对比进一步说明本发明的有益效果:
[0028]
附图说明
[0029]
图1:3-n-乙酰依替米星质谱图
[0030]
图2:3-n-乙酰依替米星氢谱
[0031]
图3:3-n-乙酰依替米星碳谱
[0032]
图4:-乙酰依替米星hsqc谱
[0033]
图5:3n-乙酰依替米星cosy谱
[0034]
图6:3-n-乙酰基依替米星薄层色谱图
[0035]
图7:3-n-乙酰依替米星粗品制备图
[0036]
图8:3-n-乙酰依替米星检测图
具体实施方式:
[0037]
以下通过实施例进一步说明本发明,但不作为对本发明的限制。
[0038]
实施例1:
[0039]
取庆大霉素c1a 20.0g溶于甲醇200ml中得溶液1。溶液1中加入无水乙酸锌20.6g,搅拌溶解搅拌2小时,得反应液1。
[0040]
反应液1冷却至0~10℃,加入三乙胺20ml和乙酸酐18.0ml,搅拌2小时,得反应液2。
[0041]
反应液2浓缩去溶剂,加入水300ml溶解,加入草酸钠16.6g,搅拌3小时,沉淀过滤,得滤液2。滤液浓缩去溶剂,以大孔树脂分离,得到中间体p1。
[0042]
中间体p1加入乙二醇二甲醚100ml、六甲基二硅氮烷120ml和浓硫酸0.2ml,90℃加
热回流4小时,得反应液3。
[0043]
反应液3浓缩蒸去大部分溶剂,以二氯甲烷5.4ml溶解,得溶液3。溶液3冷却至0~10℃,加入40%乙醛4.8ml,搅拌1小时,得反应液4。
[0044]
反应液4中加入硼氢化钾7.2g,搅拌1小时,得反应液5。
[0045]
反应液5中加入ph为10的硼酸缓冲液30ml(由10.0g硼酸加去离子水30ml搅拌溶解,用氢氧化钠调节ph=10制得),搅拌5小时,得反应液6。
[0046]
反应液6中加入10%氢氧化钠溶液100ml,浓缩蒸去二氯甲烷,再加入20%氢氧化钠240ml溶液,升温至110℃回流,搅拌3小时,得反应液7。
[0047]
反应液7以大孔树脂吸附,以无盐水冲洗除盐,以5%~25%乙醇梯度洗脱,收集纯度>80%的洗脱液,真空浓缩后得到粗品,
[0048]
粗品按照以下制备液相条件进行分离纯化:
[0049][0050][0051]
经制备液相分离得3-n-乙酰依替米星纯品,共收集1.2g。
[0052]
综合解析:
[0053]
3-n-乙酰依替米星exact mass为519.3268,分子式为c
23h45
n5o8;在高分辨质谱正离子模式下,显示准分子离子峰为520.33417,即为([c
23h45
n5o8]+h)
+
,在负离子模式下,准分子离子峰为518.31812,即为([c
23h45
n5o8]-h)-,均与3n-乙酰依替米星的分子式相符。
[0054]
在nmr图谱中,本品的1h nmr中共有20组峰(除溶剂峰外),其积分比从低场到高场为1:1:1:1:2:2:1:1:1:1:1:3:3:5:3:3:1:3:5:6,共45个质子;
13
c nmr谱图中从低场到高场有23个c信号(除溶剂峰外),因此共含有23个碳原子;同时通过hsqc和cosy分析,样品与3n-乙酰依替米星结构式相符且能得到合理的解释。
[0055]
综上所述,本品通过hrms和nmr的方法,确证了样品与3n-乙酰依替米星结构式一致。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1