一种氧化型谷胱甘肽及其晶型和杂质的制备方法与流程
发明领域本发明属于化学合成领域,具体涉及氧化型谷胱甘肽及其七水合物晶型和杂质的制备方法。
背景技术:
1、氧化型谷胱甘肽与还原型谷胱甘肽的作用类似,但氧化型谷胱甘肽的稳定性优于还原型谷胱甘肽,所以可替代还原型谷胱甘肽,用作保健食品、医药、化妆品等制品的有效成分。
2、目前已报道的氧化型谷胱甘肽制备方法全是由还原型谷胱甘肽氧化而成,例如:
3、路线一:过氧化氢氧化法(中国医药工业杂志,2013,44,265)。该方法是将还原型谷胱甘肽溶于水中,调节至合适的ph值后,以过氧化氢作为氧化剂对谷胱甘肽进行氧化,从而制得氧化型谷胱甘肽。该方法的缺点在于:过氧化氢氧化反应的速度虽然相对较快,但反应也较为激烈,所以反应条件比较苛刻,需要严格地控制反应体系的温度、ph、过氧化氢的用量等工艺参数,否则会出现较多的过氧化杂质或者降解杂质,影响产品纯度和收率。另外,氧化剂过氧化氢在我国属于易制爆危险化学品,使用须受到管控。此外,过氧化氢还容易发生自降解,需要密闭低温保存。
4、路线二:精氨酸催化法(rsc adv.,2014,4,33399–33407)。该方法将还原型谷胱甘肽溶于水中,以氧气作为氧化剂,精氨酸为催化剂,反应无废料产生,绿色环保,而且氧气和精氨酸比较容易获得,原料获取比较方便。该方法的缺点在于:反应过程中需要使用精氨酸作为催化剂,后处理过程中容易有残留的精氨酸;而且反应需要加热到50℃,产物氧化型谷胱甘肽在高温条件下容易发生部分降解和消旋化,导致收率降低并且纯度下降。
5、路线三:酶催化法(日本特开平5-146279号公报)。该方法属于生物酶催化法,需要使用特定的生物氧化酶做催化剂,利用空气将还原型谷胱甘肽水溶液氧化,得到氧化型谷胱甘肽。该方法的缺点在于:酶的获取和保存比较困难,不如常用的化学试剂。而且反应结束后还要将酶从反应液中分离出来,需要特定的工艺技术和设备,一般化工原料药企业的现有设备可能不适用。
6、路线四:溴代丙二酸二乙酯法(chem.pharm.bull.1986,34,486-495)。该方法将还原型谷胱甘肽溶于碱性的水/乙醇溶液,然后将溴代丙二酸二乙酯的乙醇溶液滴加进去,在-16℃反应1个小时。该方法的缺点在于:氧化剂使用溴代丙二酸二乙酯,成本相对较高,而且溴代丙二酸二乙酯生成的副产物较多,操作复杂。反应过程需要-16℃的低温,相对不适宜工业化。
7、此外,通过上述方法制备的氧化型谷胱甘肽的晶型主要包括非结晶无定型体(cn102858792a)、一水合物晶体(日本特许第4401775号公报)、六水合物晶体(cn102869674a)和八水合物晶体(international union of crystallography,pp538,1999)。其中非结晶无定型体水溶性较差限制了其在医药行业的应用;一水合物晶体由于其为针状晶体且易结块,所以晶体分离能力差,而且其中杂质难去除;六水合物晶体在结晶过程中需要调节ph,且析晶时间长达十几个小时,操作较繁琐,得到困难;八水合物晶体的水含量不均匀、稳定性差且需要长达3-4天的长时间来获得晶体。而且,氧化型谷胱甘肽现有的工艺都没有实现大规模的工业化生产,主要还是在于反应的温和性(减少产物降解与消旋)与经济性难以同时实现,需要一个既温和又经济易得而且副产物少的氧化体系和结晶体系,以实现优异的工艺稳定性。
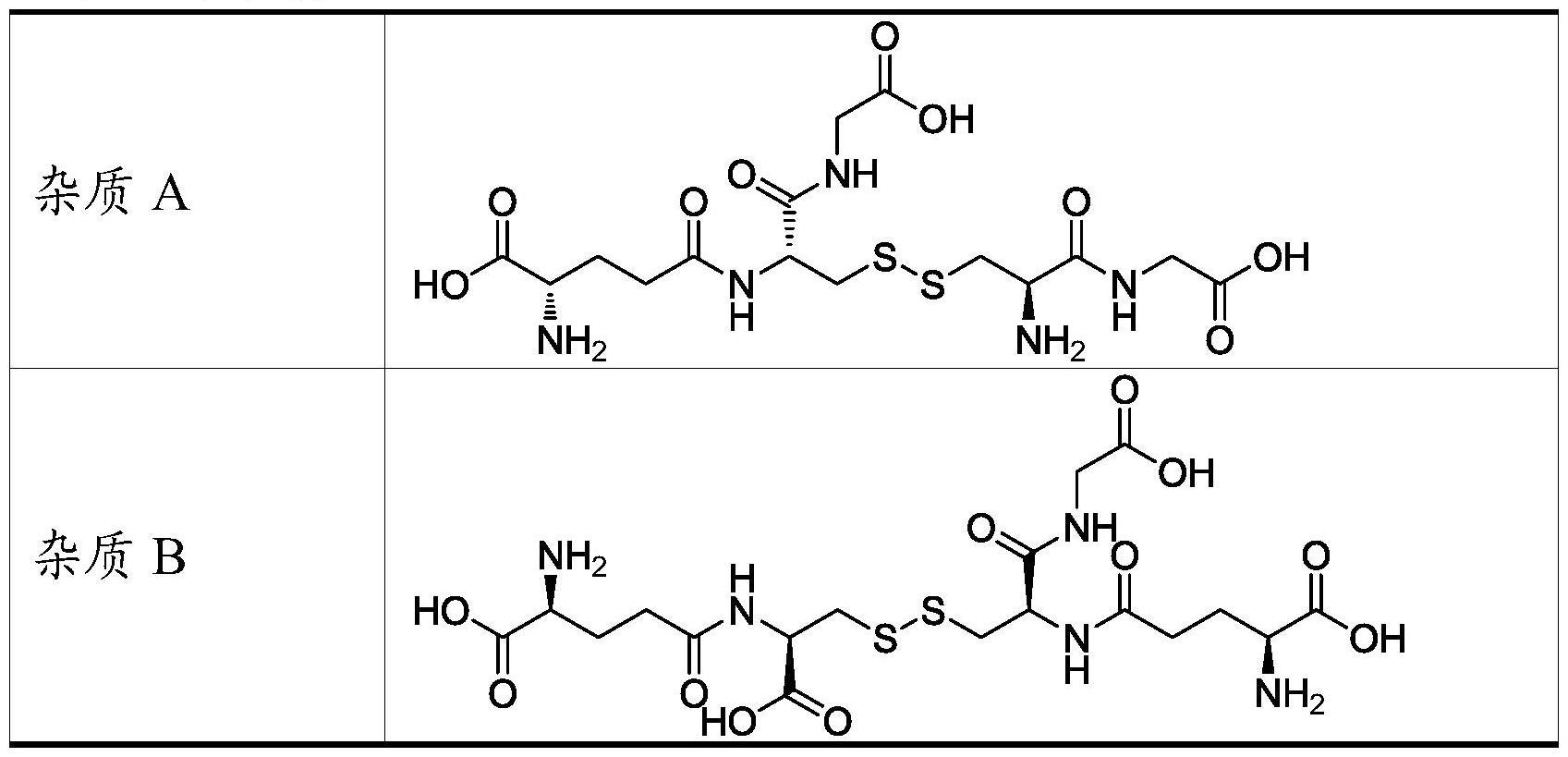
8、此外,经过液相色谱的检测以及质谱的结构确证,氧化型谷胱甘肽中会含有以下三种杂质:
9、
10、
11、有关这三种杂质的制备方法报道较少,且所用原料、试剂无法在市场上购得,缺少实用价值。
12、关于氧化型谷胱甘肽的精制过程没有文献报道,多肽类化合物因为易水解、易消旋、容易生物降解等原因,常用的纯化精制方法为使用离子交换树脂色谱分离或者使用制备液相分离等方法。以上方法的溶剂损耗较高,生产成本较大。
13、综上,本领域对于具有成本低、反应条件温和、产物纯度高、适用于工业化生产的氧化型谷胱甘肽的合成方法存在需求。
技术实现思路
1、相对于现有方法,本技术提供的氧化型谷胱甘肽的合成方法能够解决以上问题。
2、具体地,本发明涉及以下技术方案:
3、1.式(i)化合物的七水合物晶体:
4、
5、2.技术方案1的晶体,其特征在于:在使用cukα辐射得到的x射线粉末衍射图中,至少包括位于以下°2θ表示的特征峰:8.238±0.2、16.338±0.2和24.551±0.2。
6、3.技术方案2的晶体,其特征在于:在使用cukα辐射得到的x射线粉末衍射图中,还至少包括位于以下°2θ表示的特征峰:10.619±0.2、19.539±0.2、26.806±0.2和34.618±0.2。
7、4.技术方案3的晶体,其特征在于:在使用cukα辐射得到的x射线粉末衍射图中,还至少包括位于以下°2θ表示的特征峰:9.750±0.2、22.738±0.2和23.200±0.2。
8、5.技术方案1的晶体,其特征在于:其具有基本上如图5所示的x射线粉末衍射图。
9、6.技术方案1-5中任一项的晶体,其特征还在于:其具有169±2℃的熔点。
10、7.技术方案1-6中任一项的晶体,其特征还在于:在热解重量分析中,其在50-100℃失重约14±1%,在100-160℃失重约3±1%。
11、8.技术方案1-7中任一项的晶体,其特征还在于:其热解重量图如图6所示。
12、9.一种制备式(i)化合物的方法,
13、
14、包括将式(ii)化合物用dmso处理,氧化成为式(i)化合物。
15、10.技术方案9的方法,其中dmso与式(ii)化合物的摩尔比为2:1至25:1,优选2.5:1至5:1,更优选2:1、2.5:1、3:1、3.5:1、4:1、4.5:1或5:1。
16、11.技术方案9或10的方法,其中额外向反应中加入极性溶剂,促进式(ii)化合物溶解;优选地,所述极性溶剂选自水、甲酰胺、三氟乙酸、乙腈、dmf、六甲基磷酰胺、甲醇、乙醇、乙酸、异丙醇、吡啶、四甲基乙二胺、丙酮、三乙胺、正丁醇、二氧六环、四氢呋喃、甲酸甲酯、三丁胺、甲乙酮、乙酸乙酯、氯仿、三辛胺、碳酸二甲酯和乙醚,或其化合物,更优选水。
17、12.技术方案11的方法,其中所述极性溶剂与式(ii)化合物的比例为每100g式(ii)化合物添加200至2000ml极性溶剂,优选地250ml至1000ml,更优选250ml、300ml、500ml、750ml或1000ml。
18、13.技术方案9-12中任一项的方法,其中将反应的ph调节至2至9,优选3至7,更优选5至7,例如5、5.5、6、6.5、7、8或9。
19、14.技术方案9-13中任一项的方法,其中将反应在-10℃至60℃进行,优选5℃至30℃,例如25℃、40℃或室温。
20、15.技术方案9-14中任一项的方法,其中所述反应进行5至60h,优选地10至48h,例如10h、15h、20h、25h、30h、35h、40h或45h。
21、16.技术方案9-15中任一项的方法,其中在反应结束后,将其ph调节至氧化型谷胱甘肽的等电点(ph 2.75至2.90),搅拌析晶至少5h,优选地至少10h。
22、17.技术方案16的方法,其中所述搅拌析晶在5℃至40℃,优选5℃、10℃、15℃、20℃或室温进行。
23、18.技术方案9-17中任一项的方法,其中所得式(i)化合物的纯度≧98%,优选地≧98.5%,优选地≧99%,优选地≧99.7%,优选地≧99.8%,优选地≧99.9%;并且其中产物中杂质a、杂质b和杂质c的总含量低于1%,优选低于0.5%,优选低于0.3%,优选低于0.1%,
24、
25、19.一种精制技术方案9-18中任一项的氧化型谷胱甘肽的方法,包括:
26、1)将氧化型谷胱甘肽的粗品溶于纯化水中,纯化水用量为氧化型谷胱甘肽粗品质量的3~5倍;
27、2)溶解后趁热过滤;
28、3)滤液降温至10~25℃,优选12~20℃析晶。
29、20.技术方案19的方法,其中在步骤1)中,纯化水的温度为35℃~55℃,优选为40℃~50℃;并且任选地加入氯化亚铁溶液以更好的去除残留的原料,优选地,每公斤氧化型谷胱甘肽加入100ml 5%氯化亚铁溶液。
30、21.技术方案19或20的方法,其中在步骤3)中,重结晶的析晶时间为3~10小时,优选4~6小时;优选地,采用梯度降温,降温速率为每小时10℃。
31、22.一种制备技术方案1-8中任一项的氧化型谷胱甘肽七水合物晶体的方法,包括将技术方案9-21中任一项制备的氧化型谷胱甘肽在纯化水中进行重结晶;优选包括以下步骤:
32、1)将氧化型谷胱甘肽溶于纯化水中,搅拌至溶解;
33、2)溶解后趁热过滤,然后梯度降温至目标温度;
34、3)控温析晶。
35、23.技术方案22的方法,其中在步骤1)中,每公斤氧化型谷胱甘肽使用2l至6l的纯化水,优选3l至6l、更优选3l至5l、最优选4l的纯化水。
36、24.技术方案22或23的方法,其中在步骤1)中,溶解过程中纯化水的温度为40-60℃,优选40-50℃,更优选50℃。
37、25.权利要求22-24中任一项的方法,其中在步骤1)中加入氯化亚铁溶液以更好的去除残留的原料,优选地,每公斤氧化型谷胱甘肽加入100ml5%氯化亚铁溶液。
38、26.技术方案22-25中任一项的方法,其中在步骤2)中,降温梯度为5-25℃/h,优选5-20℃/h,优选5-15℃/h,更优选10℃/h。
39、27.技术方案22-26中任一项的方法,其中步骤2)中的目标温度为5-25℃,优选10-25℃,更优选15-25℃。
40、28.技术方案22-27中任一项的方法,其中步骤3)中的温度控制在5-25℃,优选10-25℃,更优选15-25℃。
41、29.技术方案22-28中任一项的方法,其中步骤3)中的析晶时间为6-12小时,优选6-8小时。
42、30.化合物,其选自:
43、
44、
45、31.一种制备技术方案30中的杂质a的方法,
46、
47、包括将摩尔比1:1的还原型谷胱甘肽和cys-gly用dmso氧化。
48、32.技术方案31的方法,其中其中dmso的用量为2~10当量,优选3~5当量。
49、33.技术方案31或32的方法,包括以下步骤:
50、1)将摩尔比范围为1:1~1:3的boc-cys(trt)-oh和甘氨酸叔丁酯在1.5~4当量的缩合剂(如hatu、hbtu、pybop、depbt等,优选hbtu和depbt)和2当量的有机叔胺(如n,n-二异丙基乙胺和三乙胺,优选n,n-二异丙基乙胺)催化下进行缩合反应,其中所述反应在dmf或二氯甲烷中进行;
51、2)用三氟乙酸脱去trt、boc和tbu保护基,得到cys-gly;
52、3)将摩尔比1:1的还原型谷胱甘肽和cys-gly用3~5当量的dmso氧化。
53、34.一种制备技术方案30中的杂质b的方法,
54、
55、包括将摩尔比1:1的还原型谷胱甘肽和glu-cys用dmso氧化。
56、35.技术方案34的方法,其中dmso的用量为2~10当量,优选3~5当量。
57、36.技术方案34或35的方法,包括以下步骤:
58、1)将摩尔比范围是1:1~1:2的boc-glu-otbu和h-cys(trt)-otbu在1.5~4当量的缩合剂(如hatu、hbtu、pybop、depbt等,优选hbtu和depbt)和2当量的有机叔胺(如n,n-二异丙基乙胺和三乙胺,优选n,n-二异丙基乙胺)催化下进行缩合反应;
59、2)用三氟乙酸脱去boc、trt和tbu保护基,得到glu-cys;
60、3)将摩尔比1:1的还原型谷胱甘肽和glu-cys用3~5当量的dmso氧化。
61、37.一种制备技术方案30中的杂质c的方法,包括将摩尔比1:1的还原型谷胱甘肽和半胱氨酸用dmso氧化。
62、38.技术方案37的方法,其中dmso的用量为2~10当量,优选3~5当量。
63、本发明的有益效果:
64、该方法得到的氧化型谷胱甘肽粗品中不含有过氧化杂质,水解杂质含量更低,且没有其他固体副产物生成;对其他杂质的结构给予分析和合成后,对后续氧化型谷胱甘肽的质量控制提供了依据和保证。
65、本发明偶然发现,在氧化型谷胱甘肽的精制过程中,加入少量氯化亚铁水溶液,意想不到的可以更好的提高纯化效果,增加产物纯度;而且采用一定的条件在纯化水中进行重结晶,可以得到氧化型谷胱甘肽七水合物晶体。
- 还没有人留言评论。精彩留言会获得点赞!