美托咪定及其衍生物的制备方法与流程
本发明涉及药物化学领域,并描述了制备美托咪定及其衍生物的方法。
背景技术:
1、按美托咪定是一种合成药物,是4-[1-(2,3-二甲基苯基)乙基]-1h-咪唑的两种立体异构体的外消旋混合物。美托咪定是一种强效的α2-肾上腺素能受体激动剂,具有剂量依赖性镇痛和/或镇静作用。通过作用于无脊椎动物的g蛋白偶联章鱼胺受体,美托咪定可抑制软体动物的附着,用于防止海洋船舶水下船体的生物污垢。
2、所描述的大多数生产美托咪定的方法都是基于使用已经含有咪唑环的前体,例如4(5)-咪唑羧酸甲酯(us4544664a,1985)、n-(三甲基硅基)咪唑(wo2009/53709,2009;cn105254567,2016;cn106749027,2017;cn106588777,2017;cn111253316,2020和cn112979552,2021)、4(5)-乙酰基咪唑(cn105884691,2016)、4(5)-碘咪唑(cn106083724,2016)、(1-三苯甲基-1h-咪唑-4-基)硼酸(cn108147999,2018)、4(5)-咪唑甲醛(cn107857731,2018和kr2021/12112,2021)、1-二甲基氨基磺酰基-2-(叔丁基二甲基硅基)咪唑(cn109608400,2019)和1h-咪唑-4-甲腈(wo2021/89878,2021)。
3、然而,上述前体的制备是单独的多阶段方法,通常需要特殊的反应条件,因为这些化合物在大多数情况下不可商购。
4、特别地,专利cn109608400描述了在-78℃下与正丁基锂反应以获得关键前体(1-二甲基氨磺酰基-2-(叔丁基二甲基甲硅烷基)咪唑),其需要使用低温低温单元和专门设计的反应器。
5、在专利cn108147999中描述的制备美托咪定的方法中使用的(1-三苯甲基-1h-咪唑-4-基)硼酸也不是市售的前体。其制备是单独的多阶段工艺流程。
6、基于n-(三甲基甲硅烷基)咪唑与1-(1-氯乙基)-2,3-二甲基苯(cn105254567,2016;cn106749027,2017;cn111253316,2020和cn112979552,2021)或1-(2,3-二甲基苯基)乙醇(wo2009/53709,2009和cn106588777,2017)通过friedel-crafts反应的相互作用的美托咪定合成方法的缺点是使用大量过量的路易斯酸(本专利中的四氯化钛)。
7、制备美托咪定的第二种合成方法是从最初不含咪唑的前体产生咪唑循环。在这种情况下,作为规则,使用线性而不是收敛合成方案,并且通常在最后阶段产生咪唑环。特别地,在本发明中描述了与美托咪定生产有关的这种方法。
8、目前,仅有很少的文献来公开与美托咪定的生产有关的这种方法。具体地,专利fi77858c(1982)、wo2011070069a1(2011)、wo2012/172122(2012)、wo2012/172120(2012)、wo2013/14428(2013)、wo2016/120635(2016)、cn106518812(2017)和cn111217756(2020)。
9、在专利fi77858c(1982)中描述了用于获得美托咪定的第一种方法之一,包括咪唑环的产生,并且其基于bredereck反应(hellmut bredereck.chem.ber.1953,86,88)。尽管其收率适中(最后阶段约30%),但反应容易进行并且不需要特殊条件。然而,该专利没有描述用于合成引入bredereck反应的关键前体的任何方法,而是基于在专利fi77858c的公开时间已知的α-卤代酮的合成方法(不包括直接卤化),最广泛使用的方法是基于重氮甲烷与羧酸卤化物的相互作用,随后是所得重氮酮与相应的卤化氢的一锅式反应(nierensteinm.重氮甲烷对一些芳族酰氯的作用.j.chem.soc.trans.1915,107,1491-1494)。由于重氮甲烷和重氮酮是极具爆炸性的并且也是有毒的化合物,因此使用该技术生产美托咪定具有严重的缺点,特别是当工艺按比例增加时发生高爆炸危害。
10、在我们的观点中,排除了该前体作者通过直接卤化相应的羰基化合物的制备,因为迄今为止在文献中没有描述合成3-(2,3-二甲基苯基)-2-丁酮7的实际方法。其中3-(2,3-二甲基苯基)-2-丁酮7被提及为由1,5,5,8-三甲基-6-亚甲基-三环[3.2.1.02.7]辛-3-烯-8-内型醇的重排产生的异构酮的复杂、难以分离的混合物的组分的唯一公开是gabrielemukheree-méler等人的论文。(helv.chim.acta 1977,60,1758–1780).
11、专利wo2011070069a1(2011)公开了在从市售的2-二甲基苯甲酸开始的多步骤工艺中产生咪唑环的合成方法。然而,该方法具有显著的缺点,即需要在压力下进行氢化反应(该合成中的氢化反应分两个阶段进行),需要使用专用设备(钢反应器)以及气态氢。
12、专利wo2013/14428(2013)和wo2016/120635(2016)描述了基于2-溴-3-(2,3-二甲基苯基)丁醛与超过25%氨水和乙醇的乙酸甲脒的缩合制备美托咪定的方法。所述方法涉及很少的步骤,实用性强,但也需要在压力下反应。
13、wo2012/172122(2012)和wo2012/172120(2012)专利中公开的生产美托咪定的方法,包括创建咪唑环,该方法根据van loysen反应基于2-(2,3-二甲基苯基)-1-丙醛与甲苯磺酰基甲基异氰酸酯的相互作用((a)albert m.van leusen.tetrahedron lett.1972,2369-2372;(b)j.org.chem.1977,42,7,1153-1159)具有很大的缺点和局限性。特别是需要对美托咪定碱进行色谱纯化,以及使用有毒的氰化钠。此外,与上述专利wo2013/14428(2013)类似,导致形成咪唑环的反应也是在压力下进行的。
14、专利cn106518812(2017)中描述的美托咪定合成方法也是基于van loysen反应,与上述专利wo2012/172122(2012)和wo2012/172120(2012)具有类似的缺点。
15、因此,目前有必要开发生产美托咪定的新型合成路线,这种路线在引入工业生产时将更加安全且相当便宜。我们开发的合成路线的优势在于可以对最终产品(美托咪定)的分子进行改性,并获得各种具有生物活性的衍生物。
16、此外,我们声称的生产美托咪定及其衍生物的新方法与专利wo2013/14428(2013)和wo2016/120635(2016)中公开的方法相比,在以下参数上更胜一筹:
17、1.3-(2,3-二甲基苯基)-2-丁酮7(美托咪定的关键前体)的合成使用一锅法进行,其中交叉偶联反应使碳骨架显著复杂化,并且保护基团去除在一个阶段进行,这大大减少了合成步骤的总数。
18、
19、2.中间体1-溴-3-(2,3-二甲基苯基)丁-2-酮5,3-(2,3-二甲基苯基)-2-氧代丁基乙酸酯4和n-[3-(2,3-二甲基苯基)-2-氧代丁基]-n-甲酰基甲酰胺22无需进一步纯化即可用于后续步骤。
20、
21、3.由于所描述的合成方法是基于咪唑环的产生,因此不需要使用已经包含这种环的难以到达的前体。
22、4.在所提出的制备美托咪定的方法中描述的所有反应都在大气压下进行,这消除了对专用设备例如高压反应器的需要。
23、5.所公开的用于生产需要冷却或加热的美托咪定的方法中描述的任何反应在中等低(-15℃)或中等高温(≤130℃,路线a和b,以及≤160℃,路线c)下进行,这消除了对专用设备诸如低温低温系统和特别设计的反应器的需要。
技术实现思路
1、根据本发明的方法在以下反应方案中公开:
2、方案1.路线a
3、
4、方案2.路线b
5、
6、方案3.路线c
7、
8、制备3-(2,3-二甲基苯基)丁-2-酮7的起始化合物为市售的2,3-二甲基溴苯11
9、
10、也可根据文献方法从2,3-二甲基苯胺13
11、
12、中获得(方法改进自下文讨论的j.chem.soc.1940,16-18)。
13、反应开始于由2-二甲基苯胺13和40%氢溴酸产生2-二甲基苯胺氢溴酸盐。将未经分离和纯化的盐引入到与从亚硝酸钠原位产生的亚硝酸和过量吸收的氢溴酸的反应中。
14、由于重氮化过程放热进行,因此需要小心地控制反应器中的内部温度,而不允许其上升超过0℃,这通过改变亚硝酸钠溶液的添加速率来实现。
15、由于重氮盐12
16、
17、会发生副反应和分解,超过规定温度会导致产品的产量显著下降,这在某种程度上已经在0下观察到。
18、在2,3-二甲基苯基重氮溴化物12的溶液中加入新沉淀的铜粉,并将溶液温度升至28℃以上,使重氮盐分解,释放出气态氮,生成2,3-二甲基溴苯11。
19、在足够快的搅拌速度下进行重氮盐12的分解过程,可以避免反应混合物因氮气进化而起泡,并确保过程顺利。
20、温度自发停止上升后,将反应混合物短暂(20-30分钟)加热至70-75°с,以完成整个过程,冷却后用轻质有机溶剂如正己烷、庚烷或轻质石油醚萃取。不建议使用戊烷,因为其沸点较低,在加工和回收过程中会导致大量溶剂流失。
21、由于重氮化过程中会形成大量的2,3-二甲基苯酚25
22、
23、用氢氧化钠或氢氧化钾等强碱水溶液清洗提取物是必要的操作。通过从碱性溶液中进行蒸汽蒸馏,可将产品11与树脂杂质分离。
24、不建议使用重溶剂(二氯甲烷、氯仿)来萃取2,3-二甲基溴苯11,因为它们的密度与反应混合物的密度接近,而且在碱性介质中容易形成稳定的乳状液,这使用碱性溶液洗涤有机相以除去2,3-二甲基苯酚25杂质的过程变得非常复杂。
25、下一步中使用的结构14
26、的化合物可以根据例如greene's protective groups in organicsynhesis 2006(greene的《有机合成中的保护基团》,2006)中描述的标准程序,从市售的3-溴-2-丁酮(结构15)
27、
28、中合成。
29、起始3-溴-2-丁酮15也可以根据文献方法(e.g.,rec.trav.chim.pays-bas1946,65,691)通过在乙酸水溶液中用分子溴溴化容易获得的甲基乙基酮17
30、
31、来获得。
32、由于甲基乙基酮17的溴化反应导致形成两种区域异构体,即3-溴-2-丁酮15和1-溴-2-丁酮16
33、
34、因此在引入保护基团之前,应该通过在有效蒸馏塔上真空蒸馏来分离混合物。根据nmr,高800mm且直径30mm填充有拉式环(raschig环)的柱提供含有≤5%区域异构体16的3-溴-2-丁酮15。hplc或gc也可用于评价馏出物中区域异构体的15∶16比。
35、3-溴-2-丁酮15与乙二醇在催化量的对甲苯磺酸一水合物存在下在甲苯中的相互作用,同时共沸蒸馏反应期间形成的水,导致乙烯缩酮14的形成。
36、下一阶段包括由2,3-二甲基溴苯11制备2,3-二甲基苯基溴化镁10
37、
38、所得有机镁试剂与2-(1-溴乙基)-2-甲基-1,3-二氧戊环14反应,并除去保护基团。
39、所列反应在一锅中进行,无需分离和纯化任何中间产物,即可生成酮7
40、
41、格氏试剂10的制备首先是2,3-二甲基溴苯11和金属镁在干燥的四氢呋喃(thf)中直接反应,反应温度不超过50℃(最佳温度为48-50°с)。为了完成反应,需要短期加热(70-75℃,45-60分钟)。
42、溶剂(四氢呋喃)的预处理包括在氩气环境下进行蒸馏,并用活化的3a分子筛进行干燥。要从thf中去除氢过氧化物,可采用文献中描述的标准方法,如用活性氧化铝吸附、用dowex-1阴离子交换树脂处理以及蒸馏时初步添加对苯二酚((a)zakharov l.n.化学实验室的安全(safety in chemical laborato-ries)1991;(b)donald e.clark.过氧化物和过氧化物形成化合物(peroxides and peroxide-forming compounds).chem.healthsaf.2001,8,5,12-22).
43、溶液中2,3-二甲基苯基溴化镁10的最佳浓度为0.8-1m。在该浓度以上,溶液粘度增加,这使其在交叉耦合期间的添加过程复杂化。
44、分析配制溶液中2,3-二甲基苯基溴化镁10的浓度和产率,可通过滴定溶解在四氢呋喃中氯化锂饱和溶液中的精确重量的碘,直到溶液的棕色颜色消失为止(paul knochel.有机金属锌、镁和镧系试剂的便捷滴定法(convenient titration method for organo-metallic zinc,magnesium,and lanthanide reagents).synthesis 2006,5,890-891)。
45、通过使用gc估计起始的2,3-二甲基溴苯11的转化率,然后对获得的数据进行外推,分析溶液中有机镁试剂的浓度,由于发生副反应形成联苯9,不能被认为是评估有机镁试剂产率的正确方法
46、
47、2,3-二甲基苯基溴化镁10与2-(1-溴乙基)-2-甲基-1,3-二氧戊环14在10摩尔%乙酰丙酮钴(iii)和10摩尔%n,n,n',n'-四甲基乙二胺(tmeda)的存在下在thf中的反应导致形成新的键,并形成环状缩酮8
48、
49、环状缩酮8在酸性介质中很容易裂解(10%盐酸,水解速度小于5分钟,uplc监测),这使得去保护过程与淬灭反应混合物同时进行成为可能。
50、对交叉偶联反应进行优化后发现,催化剂用量从10摩尔%降至5摩尔%,会导致酮7的产率从87.2%降至60%。
51、用非极性溶剂(庚烷)萃取并在真空中浓缩后,酮7中含有大量(根据核磁共振数据,≥20%)乙二醇,乙二醇是保护基裂解后形成的,与产物一起在真空中蒸馏,这导致蒸馏温度范围扩大到10℃,从而降低了产率,因为在开始收集主要馏分之前,需要分离出更多的早期馏分。
52、在真空蒸馏之前,通过用水反复洗涤3-(2,3-二甲基苯基)丁-2-酮7,可以去除其中的乙二醇。由于酮7的密度接近水的密度,洗涤过程因形成稳定的乳状液而变得复杂。用正己烷或其他轻质非极性溶剂(~1:1.5vol.)对粗酮7进行初步稀释,可以完全避免在洗涤过程中形成任何乳状液。乙二醇杂质是否完全去除,可通过核磁共振光谱(亚甲基信号消失(3.76ppm,s,cdcl3))或气相色谱法来控制。
53、交叉偶联反应中形成的第二种副产物是2,2',3,3'-四甲基联苯9,它也必须在蒸馏过程之前分离出来,因为它在真空中易挥发,是蒸馏过程结束时目标酮7的污染源。
54、用甲醇(体积比为1:1)稀释粗酮7,然后在-15...-20℃温度下结晶联苯92天,可以有效地去除大部分2,2',3,3'-四甲基联苯(hplc检测结果大于95%)。将结晶化合物9作为晶种引入从残留物中得到的溶液中,可将结晶时间缩短至约20小时。
55、在非极性有机溶剂(如己烷)中,用粒度为40-63μm的活化硅胶吸附钴化合物(120℃,20小时),可去除痕量的钴化合物。
56、在下一阶段,生成的3-(2,3-二甲基苯基)-2-丁酮7在末端甲基上用分子溴溴化,形成1-溴-3-(2,3-二甲基苯基)-2-丁酮5
57、
58、提供最高区域选择性的最佳溶剂是甲醇。溴化过程在宽的温度范围(从-15℃至室温)内进行,然而,在反应期间形成的副产物(二溴衍生物和三溴衍生物)的量以及其速率极度取决于溶液中起始酮的温度和浓度。
59、室温下的溴化(20-25℃)已经在加入溴的瞬间进行,并在几分钟内结束。然而,利用这种溴化方式,多溴化产物的杂质的量大大增加(目标产物的hplc面积5≤49%)。
60、当在-12℃溴化时,多溴杂质的形成速率显著减慢(hplc面积为581%,二溴酮的hplc面积<5%),然而,达到起始酮的转化率>95%(hplc)需要约28小时。
61、溴化的最佳温度范围为-5至-8℃。
62、溴∶酮7的化学计量比的使用导致后者仅≤85%转化率(hplc)。为了实现起始酮的完全转化,需要至少1.15当量的溴,最佳地1.20-1.25当量的溴。
63、通过溶液的稀释程度也可以实现对侧工艺过程的控制。每1g酮使用少于10ml的溶剂导致伴随多溴化杂质形成的副反应的急剧增加。酮7在溶液中的最佳浓度在0.18-0.22mol/l的范围内。
64、向酮的甲醇溶液中加入溴应该尽可能快地进行,优选在一部分中进行,因为通过缓慢加入或逐滴加入,形成显著量(hplc面积>10%)的区域异构溴化产物20的杂质
65、
66、要完成反应,可能需要将温度升至0°с(约17-18小时后),并保持在该温度下,直到溴色完全消失。如果起始酮7的转化不完全,可能需要引入额外的溴部分,这样可以使化合物7的转化率大于97%(hplc)。
67、在溴化过程中形成的1-溴-3-(2,3-二甲基苯基)丁-2-酮二甲基缩醛6
68、
69、在反应混合物中含有过量的溴化氢,在用水稀释后很容易在一锅中裂解,无需分离(ph值为3时的水解速率小于2分钟,uplc),形成1-溴-3-(2,3-二甲基苯基)丁-2-酮5,无需进一步纯化即可用于下一步。产品5在室温下不稳定,颜色很快变深,但可在-18℃下保存数周而不会明显降解。
70、使用乙酸、酯类(如乙酸乙酯)或卤代烃作为溶剂,主要会生成取代度最高的3-溴-3-(2,3-二甲基苯基)丁-2-酮20。
71、在四氢呋喃、乙酸乙酯或冰醋酸中使用吡啶氢溴酸过溴化物(phbp,19)作为溴化剂,也几乎只能生成3-溴-3-(2,3-二甲基苯基)丁-2-酮20,以及少量多溴化合物杂质。
72、3-(2,3-二甲基苯基)-2-氧代丁基乙酸酯(结构4)
73、
74、是形成美托咪定分子中咪唑环的前体,其合成路线为a(方案1),制备方法是在室温下,将1-溴-3-(2,3-二甲基苯基)丁-2-酮5与乙酸钾(~3eq)在合适的有机溶剂(乙腈、dmf或dmso)中反应。
75、在使用低沸点溶剂(如乙腈)的情况下,只需在真空中浓缩溶液,并对溴化钾沉淀和过量乙酸钾进行预过滤,即可从反应混合物中分离出化合物4。
76、将化合物4在非极性溶剂(正己烷、庚烷、mtbe、乙酸乙酯等)中的溶液用水洗涤,然后浓缩,即可除去乙腈。得到的3-(2,3-二甲基苯基)-2-氧代丁基乙酸酯4纯度很高,可用于下一阶段的合成,无需再提纯。
77、在处理反应混合物过程中再生的乙腈(大于90%)可以在下一次加料(合成的这一阶段)时使用,无需额外制备,不会降低产品4的质量。
78、室温下在甲醇中进行反应,起始的1-溴-3-(2,3-二甲基苯基)-2-丁酮的转化率较低(43.5小时内转化率小于60%)。
79、在使用甲醇作为溶剂以提高转化率的情况下,提高反应温度会导致副反应急剧增加,从而产生一种不适合在下一阶段使用而无需额外纯化的产物。
80、在dmf或dmso中进行反应的优点是反应时间从18-20小时(乙腈)缩短到5-6小时(dmf);但在这种情况下,需要用大量的水(3-5体积)稀释反应混合物以分离产物,然后用低沸点有机溶剂(己烷、庚烷、mtbe、乙酸乙酯等)萃取产物。
81、该过程的最后阶段包括形成一个咪唑环,并形成美托咪定的铜(i)络合物3
82、
83、分离美托咪定的游离碱2
84、
85、去除铜痕量,得到药学上可接受的盐,例如盐酸美托咪定1
86、
87、根据weidenhagen反应(weidenhagen r.chem.ber.1935,68,1953),3-(2,3-二甲基苯基)-2-氧代丁基乙酸酯4在水-醇溶液中与甲醛、氨和乙酸铜(ii)相互作用,生成美托咪定3的铜(i)络合物。
88、进行该反应的最佳助溶剂是正丙醇,它能使起始的3-(2,3-二甲基苯基)-2-氧代丁基乙酸酯4在氨水溶液中完全溶解。正丙醇的最佳用量为每1克3-(2,3-二甲基苯基)-2-氧代丁基乙酸酯4 10至15毫升。在这种情况下,美托咪定的铜(i)络合物在反应过程中会以形成良好结晶沉淀的形式预沉淀。
89、如果使用较小体积的正丙醇,会导致化合物4溶解不完全,从而形成乳状液,进而导致美托咪定铜络合物在反应器壁上和搅拌器上以致密沉淀的形式析出,使过滤过程复杂化。此外,由于反应是在液滴表面而不是在均匀介质中进行的,起始化合物4的转化不完全,因此产物的产量下降,并受到未反应前体的污染。
90、由于络合物3的溶解性,使用较大体积的正丙醇是不可行的,这可能导致其产率下降。反应在溶液的沸点(62-72℃)进行,2小时后结束。缩短加热时间(小于1.5小时)会导致络合物3铜的产率从80.9%下降到58.5%。使用甲醇、乙醇或异丙醇作为助溶剂也会导致美托咪定铜络合物在反应器壁上沉淀,这是因为它们对化合物4的溶解能力不足。
91、从铜络合物中分离游离咪唑的传统方法是在硫化氢气体的作用下将铜沉淀为硫化铜(i)。然而,由于硫化氢的毒性很高,这种方法不能被视为一种可扩展的、工业上可接受的工艺。
92、我们公开的方法避免了硫化氢的使用,而是基于无毒螯合剂的使用。
93、最佳螯合剂是二乙烯三胺五乙酸五钠盐(na5dtpa)。在这种情况下,金属与螯合剂的最佳摩尔比在1:1.2-1.5之间。
94、二乙烯三胺四乙酸四钠盐(na4edta)对美托咪定的铜(i)络合物的螯合能力不足,这导致即使使用3-5倍摩尔过量的螯合剂和较长的反应时间(>4天),起始络合物的转化率也很低,美托咪定碱的释放也不完全。
95、该过程的平衡转移也是通过将美托咪定碱不断萃取到有机相中实现的,由于铜络合物晶体表面的产物不断被清除,从而促进了反应的进行。为了加速反应,必须确保各相充分混合。
96、使用游离酸(例如edta或dtpa)及其不完全取代的盐(例如乙二胺四乙酸的二钠盐(na2edta))作为螯合剂,将美托咪定2的游离碱从铜络合物3中分离出来,会形成树脂状的络合物产物,这可能是由于游离羧基同时与咪唑环的碱性氮原子和铜离子相互作用的缘故。
97、将美托咪定碱2与盐酸等酸在合适的有机溶剂中反应,可生成盐酸美托咪定1等药学上可接受的盐。在丙酮中使用浓盐酸(35-38%)时,由于盐酸美托咪定1在水中的溶解度较高,可能会出现产品结晶的问题。在这种情况下,为了启动结晶,必须添加少量纯盐酸美托咪定晶体作为种子。在盐形成阶段加入种子会对结晶产生有利影响,结晶会在加入浓盐酸后立即或几乎立即发生。盐酸美托咪定的产率可从56.2%提高到60-65%(按两个阶段计算),方法是在过滤掉晶体的主要部分后浓缩母液(浓缩到初始体积的约1/3),然后将水-丙酮溶液在冷冻室中冷却20-25小时。
98、在形成铜络合物的阶段用任何其他醛代替甲醛,是获得咪唑环第2位有取代基的美托咪定衍生物的便捷方法。
99、另一种在美托咪定分子中形成咪唑片段的方法是路线b(方案2)。
100、在这种情况下,1-溴-3-(2,3-二甲基苯基)丁-2-酮5与二甲酰氨基钠21
101、
102、在合适的有机溶剂(如乙腈)中发生反应,得到中间体22
103、
104、无需进一步纯化即可用于下一步。
105、用于合成化合物22的市售二甲酰基酰胺钠21也可以通过文献方法从甲氧基钠和甲酰胺中以高产率获得(例如,(а)allenstein e.chem.ber.1967,100,3551;(b)us5599986a)。
106、化合物22的酸性水解导致氨基酮缩二氯化物23的形成
107、
108、该工艺的最后一步(路线b,方案2)是形成2-巯基取代的咪唑环,然后进行脱硫反应,生成美托咪定2。
109、根据marckwald反应(marckwald w.ber.dtsch.chem.ges.1892,25,2354),1-氨基-3-(2,3-二甲基苯基)丁-2-酮氢氯化物23与硫氰酸钾在水溶液中的相互作用导致形成4-[1-(2,3-二甲基苯基)乙基]-1,3-二氢-2h-咪唑-2-硫酮24
110、
111、当氨基酮盐酸盐23与过量(4-5eq)硫氰酸钾水溶液在沸点下加热时,反应会顺利进行。最佳加热时间为2.5至3.5小时。与起始试剂不同,反应产物24几乎不溶于水,并随着反应的进行从溶液中结晶出来,这大大简化了其分离过程。如有必要,可从乙醇水溶液(约65%v/v)中结晶提纯4-[1-(2,3-二甲基苯基)乙基]-1,3-二氢-2h-咪唑-2-硫酮24。
112、在酒精介质中使用雷尼镍温和地去除化合物24中的巯基,可生成高产率的美托咪定2。使用等级为w-4和w-5的雷尼镍可获得最佳效果(参见robert l.augustine.1996年《合成化学家的异相催化》中的分类(heterogeneous catalysis for the synthetic chemist1996for a classification))。
113、美托咪定分子中咪唑环的生成也可以通过1-氨基-3-(2,3-二甲基苯基)丁-2-酮盐酸盐23与甲酰胺反应一步直接完成(方案3,路线c)。尽管产率较低(约40%),但反应非常容易进行。由于甲酰胺用量过多(约10eq),在反应中既是溶剂又是试剂,而且沸点较高(210℃),因此在从反应混合物中提取美托咪定碱之前,最好先用浓氯酸(1小时,室温)水解过量的甲酰胺。
114、实验
115、缩写表
116、
117、
118、
119、通用方法
120、起始的2,3-二甲基苯胺13购自sigma-aldrich(99%,cas 87-59-2),使用时无需进一步纯化。所有使用惰性气氛的实验都是按照标准技术使用schlenk管线进行的。氩气99.999%(林德气体公司(linder gas))用作惰性气体,无需进一步净化。除thf和甲醇外,所有溶剂和试剂均购自商业供应商,使用时无需进一步纯化或制备。四氢呋喃(acs99.6%)在使用前在氩气环境中蒸馏(加入~0.1%氢醌),并在活性3a分子筛(100克/升)上干燥65小时。溴化反应中使用的甲醇在活化的3a分子筛(45克/升)上干燥168小时。分子筛在300-320℃的真空(0.01mmhg)中加热3小时后活化。为了合成有机镁反应剂,使用了fisher chem-ical公司生产的格氏反应用镁屑,并将其保存在120℃的烘箱中。根据保罗-诺谢尔(paul knochel)的方法,2,3-二甲基苯基溴化镁的分析是在氯化锂在干thf中的饱和溶液中直接用碘量滴定法进行的。在制备有机镁试剂10和交叉偶联阶段使用的部分玻璃设施(大容量反应器除外)在120℃的烘箱中干燥24小时。玻璃反应釜在处理对水分敏感的化合物之前,先在旋片泵中抽真空(0.01mmhg),在90℃下加热2小时,然后在三个抽气/充气循环中充入氩气。1h和13c nmr图谱分别在cdcl3和d6-dmso中用agilent 400mhz光谱仪(1h为400mhz,13c为100mhz)记录。化学位移以相对于所用氘化溶剂残留信号的ppm表示。重金属残留量的分析是在agilent technologies240aa原子吸收光谱仪上进行的。使用mars one微波分解系统制备重金属分析样品。70%硝酸和30%过氧化氢的混合物(体积比为10:1,180℃,25分钟)被用作样品矿化的氧化剂。tlc分析在alugram xtra sil g/uv254铝板(macherey-nagel)上进行。用254纳米紫外灯观察板面。使用带有光电二极管检测器阵列(pda)的alliance hplc系统(waters)监测反应的完整性并评估中间产物的色谱纯度。
121、分析条件
122、方法a
123、仪器:waters alliance hplc;色谱柱xbridge c18,3.5μm,4.6mm×150mm;洗脱液α:mecn;洗脱液β:h2o+0.15%vol.50% h3po4;a:b=50:50;等度洗脱模式;流速1.50ml/min;温度40℃;进样量10μl;检测波长:190-350nm。
124、方法b
125、仪器:waters alliance hplc;色谱柱xbridge c18,3.5μm,4.6mm×150mm;洗脱液α:mecn;洗脱液β:h2o+0.15%vol.50% h3po4;a:b=30:70;等度洗脱模式;流速1.00ml/min;温度40℃;进样量10μl;检测波长:190-350nm。
126、方法c
127、仪器:waters alliance hplc;色谱柱xbridge c18,3.5μm,4.6mm×150mm;洗脱液α:mecn;洗脱液β:h2o+0.15%vol.50% h3po4;梯度:0-1.20分钟30%α,1.20-12.00分钟30-90%α,12.00-15.00分钟90%α,15.01-18.00分钟90-30%α;流速1.20ml/min;温度40℃;进样量10μl;检测波长190-350nm。
128、实验
129、2,3-二甲基溴苯(11)
130、(改进自j.chem.soc.1940,16–18)
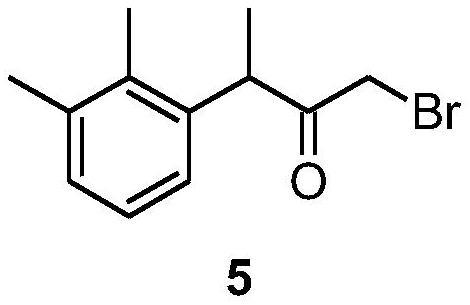
131、
132、将3,747.5克(22.23摩尔;4.0eq)48%的氢溴酸和750毫升去离子水加入一个10升的玻璃反应釜中,该反应釜配有恒温夹套和机械搅拌器,预冷至-10℃。将溶液冷却至4...5℃,然后在45分钟内从滴液漏斗中加入673.45克(5.55摩尔;1.0eq)2,3-二甲基苯胺13,搅拌(250-300转/分钟),搅拌速度应使反应混合物的温度保持在4...5℃范围内。将得到的2,3-二甲基苯胺氢溴酸盐米色晶体悬浮液冷却至-1...-2℃,然后在2小时内从滴液漏斗中加入421.8克(6.12摩尔;1.10eq)亚硝酸钠在680毫升去离子水中的溶液,加入速度应使反应混合物的温度保持在0...5℃的范围内。-2℃(反应器夹套中冷却剂的温度为-15℃)。加入亚硝酸镝溶液后,将反应混合物的温度升至0℃,并将2,3-二甲基苯基重氮溴化物12的黄棕色溶液搅拌15分钟,以完成重氮化过程。然后,提高搅拌器转速(达到450-500rpm),将100克新沉积铜粉在350毫升去离子水中的悬浮液引入反应器,并将反应器夹套中的冷却剂温度提高到30℃。当反应混合物加热到28...30℃时,重氮盐的放热分解过程开始,反应混合物的温度在4-5分钟内自发升高到60℃。自发升温停止后,反应器夹套中的冷却剂开始加热和循环,紫黑色反应混合物在70℃下加热30分钟。将反应混合物冷却至室温,用2×1,000ml轻质石油醚(40-70)萃取。下层水通过底部出口排出,深色有机相在反应器中先后用4l去离子水(10分钟)和4×4l 2.5%氢氧化钠溶液(各10分钟)洗涤,以除去2,3-二甲基苯酚。有机相分离后在真空(<40℃)下浓缩。从50克氢氧化钠与1升去离子水的溶液中蒸馏出橙红色的油,收集3.5升馏分。分离出透明的浅橙色下层,在磁力搅拌器上用100毫升浓硫酸强烈搅拌40分钟。深红色硫酸下层在分离漏斗中分离,浅黄色有机上层用2×600ml饱和碳酸氢钠溶液洗涤。水相用3×100ml亚甲基氯化物萃取。合并的有机相用无水硫酸钠干燥,过滤,真空浓缩。得到的2,3-二甲基溴苯使用300毫米真空套vigreux色谱柱进行真空分馏,收集到bp为76.2-76.5℃/6mmhg的馏分。
133、产量391.8克(38.1%)。无色液体,具有特有的二甲苯气味。1h nmr(400mhz,cdcl3)δ7.43(d,1h),7.11(d,1h),6.97(t,1h),2.40(s,3h),2.36(s,3h);13с(100mhz,сdcl3)δ138.61,136.24,130.34,128.97,126.88,125.67,21.39,19.51;tr 11.33分钟(hplc面积99.7%,方法α)。
134、3-溴-2-丁酮(15)
135、(改进自rec.trav.chim.pays-bas 1946,65,691)。
136、
137、将1,200毫升去离子水、300毫升冰醋酸和360.5克(447.8毫升;5.0摩尔)甲乙酮17注入带恒温夹套的3升玻璃反应釜中,该反应釜配有密封机械搅拌器、带压力平衡管的滴液漏斗和回流冷凝器。滴液漏斗中加入800.0克(257.9毫升;5.0摩尔;1.0等量)溴。将反应器中的溶液加热至70℃,然后在4小时内滴加溴,滴加的速度应使刚开始出现的黄色有时间随着后续溴的加入而消失。在加入溴的过程中,会形成乳状液,然后反应混合物会分离成两相。溴全部加入后,将反应混合物在70℃下搅拌30分钟,然后冷却至室温并用1升冷水(2℃)稀释。通过反应器底部出口分离出浅黄色下层,然后用3×400毫升饱和碳酸氢钠溶液(在磁力搅拌器上,在一个松散封闭的1000毫升锥形瓶中)洗涤,直到停止产生二氧化碳,再用400毫升去离子水洗涤。用无水粒状氯化钙干燥得到的15/16溴代异构体混合物(546.1克),过滤后在带抽真空夹套的800毫米高的蒸馏塔(塔内装有拉氏环)上进行真空蒸馏分离。首先分离出bp为58-60二氧化碳/38mmhg的馏分(约10毫升),然后收集bp为61-63°с/38mmhg的3-溴-2-丁酮15的主要馏分,接着收集bp为64-77℃/38mmhg的中间馏分,最后收集bp为77-78℃/38mmhg的1-溴-2-丁酮16的馏分。真空蒸馏的速率过低会导致馏出物变黄。
138、3-溴-2-丁酮(15)
139、产量279.74g(37.0%)。淡黄色液体。1h nmr(400mhz,cdcl3)δ4.37(q,1h),2.34(s,3h),1.71(d,3h);13с(100mhz,сdcl3)δ202.11,48.28,26.04,20.20。异构体1-溴-2-丁酮16的含量<5%(nmr)。tr 5.35分钟(hplc面积96.9%,方法β)。
140、1-溴-2-丁酮(16)
141、产量48.15g(6.4%)。黄色液体。1h nmr(400mhz,cdcl3)δ3.87(s,2h),2.65(q,2h),1.09(t,3h);13с(100mhz,сdcl3)δ209.92,52.65,29.13,9.35;tr 4.64分钟(hplc面积88.9%,方法β)。
142、2-(1-溴乙基)-2-甲基-1,3-二氧戊环(14)
143、
144、将259.12克(1.71摩尔)新蒸馏的3-溴-2-丁酮15、117.2克(1.88摩尔;1.10eq)乙二醇、1.58克(8.30毫摩尔;0.0048eq)对甲苯磺酸一水合物和800毫升甲苯加入2升圆底烧瓶中。烧瓶中装有一个50毫升的dean-stark捕集器,在回流条件下,反应混合物在磁力搅拌器上搅拌回流,直到接收器中不再有水滴分离出来(31毫升;3.5小时)。随着反应的进行,乙二醇的菌落层逐渐消失。无色透明溶液冷却至室温,用500毫升2%碳酸氢钠溶液和2×150毫升去离子水洗涤。分离有机层,在无水硫酸锍上干燥,过滤,在旋转蒸发仪上浓缩(35℃/10mmhg),蒸馏体积为760-780ml。产品在真空中蒸馏,收集bp为43.5-44.5℃/0.016mmhg的馏分。
145、产量271.93g(81.2%)。无色液体,无催泪作用。1h nmr(400mhz,cdcl3)δ4.05(q,1h),3.99(m,4h),1.67(d,3h),1.45(s,3h);13с(100mhz,сdcl3)δ109.96,64.74,53.22,21.22,20.65;tr 8.11分钟(hplc面积88.2%,方法β)。
146、3-(2,3-二甲基苯基)-2-丁酮(7)
147、
148、
149、将33.90克(1.39摩尔;1.05eq)镁络合物放入2升的双颈圆底烧瓶中,烧瓶在120℃的干燥箱中初步干燥过夜,并配备了一个带压力平衡臂和玻璃封装热电偶的1升滴液漏斗。向烧瓶中加入约0.8克碘,然后用热空气加热烧瓶,直到碘开始升华。将烧瓶冷却至室温后,用147毫升干燥的四氢呋喃覆盖镁,并将磁锚放入烧瓶中。将245.67克(1.32摩尔;1.0等量)2,3-二甲基溴苯11在1,000毫升干thf中的溶液装入滴液漏斗。将大约40-50毫升的溶液从滴液漏斗中一次性加入到镁葑中。从滴液漏斗中加入第一份溶液后,反应立即开始,碘色消失,温度迅速上升(<1分钟)至48℃。反应混合物温度停止上升后,立即开始从滴液漏斗中加入溶液,并积极搅拌(1300-1500转/分),搅拌速度应使反应混合物的温度保持在47-49℃范围内,而无需外部加热或冷却(约5小时)。加入所有2,3-二甲基溴苯后,将深色反应溶液在70-75℃下回流45分钟以完成反应。溶液在氩气环境中冷却。根据paul knochel的方法测定,所得溶液中2,3-二甲基苯基溴化镁10的浓度为0.99摩尔/升。2,3-二甲基苯基溴化镁的产率为92.7%。得到的组合物10溶液可直接用于下一步,无需进一步制备。
150、一个5l玻璃反应釜配有温控夹套,配备一个密封的机械搅拌器、一个热电偶和一个用于通入惰性气体的旋塞,反应釜抽空(<0.1mmhg)并在90℃温度下干燥2小时。将反应器冷却至室温并充入干燥的氩气(泵/充气循环3次)。在氩气逆流下,将41.93克(10摩尔%;117.7毫摩尔)乙酰丙酮酸钴、520毫升干thf、17.65毫升(13.67克;10摩尔%;117.7毫摩尔)tmeda和229.67克(1.177毫摩尔;1.0等量)2-(1-溴乙基)-2-甲基-1,3-二氧戊环14连续加入干燥的反应器中。悬浮液冷却至0...-1℃,在搅拌(300-400转/分钟)下,用蠕动泵以6.9-7毫升/分钟的速度向反应器中加入前一阶段制备的2,3-二甲基苯基溴化镁10溶液,持续180分钟,小心地将内部温度保持在0至-1℃(反应器夹套中的冷却剂温度为-10至-12℃,可很好地控制反应)。随着格氏试剂溶液的加入,反应混合物的颜色由绿色变为淡蓝蓝色,并从溶液中结晶出大量的溴化镁沉淀。加入全部2,3-二甲基苯基溴化镁(1.05eq)后,将反应混合物的温度升至20℃,并继续搅拌1小时。在剧烈搅拌下(400转/分),向反应混合物中缓慢加入1,460毫升9%的盐酸(2-3分钟),继续搅拌40分钟。在加入盐酸的过程中,反应混合物的颜色迅速从浅蓝蓝色变为绿色,然后变为橙色。溴化镁的结晶沉淀全部进入溶液。搅拌30分钟后,对等分的反应混合物进行hplc分析,结果显示中间酮8 100%转化为酮7。用500毫升正庚烷稀释由两相(上层淡黄色有机相和下层粉红色水相)组成的反应混合物,搅拌15分钟,然后从反应器底部出口排出下层水相。有机相依次用2×500毫升去离子水和500毫升饱和碳酸氢钠溶液洗涤。有机相用硫酸钠干燥,过滤,真空浓缩(45℃)。将得到的橙黄色油(224.0克)用240毫升甲醇稀释,并在溶液中加入100毫克2,2',3,3'-四甲基联苯9作为晶种。溶液在冷冻室(-18℃)中保存一天。用肖特漏斗过滤沉淀的2,2',3,3'-四甲基联苯9结晶悬浮液,并用2×50毫升冷甲醇洗涤。滤液在真空中浓缩;残留物用300毫升己烷稀释,并依次用1升去离子水(10分钟)、1升1%na2edta水溶液(2小时)和1升饱和氯化钠水溶液(10分钟)洗涤以除去乙二醇。利用1h nmr,通过亚甲基信号的消失(3.76ppm,s,cdcl3)来监测乙二醇去除的完整性。
151、分离有机相,用无水硫酸钠干燥,过滤,真空浓缩。使用300毫米真空夹套vigreux色谱柱对产品进行真空蒸馏。馏出物收集在珀金接收器中。弃去沸点为68-69℃/0.024mmhg的馏分,收集bp为69-72℃/0.024mm hg的主要产物馏分。
152、将得到的琥珀色3-(2,3-二甲基苯基)丁-2-酮7用250毫升己烷稀释,并用10克硅胶(40-63微米,120℃下干燥20小时)搅拌1.5小时。橙色硅胶在肖特漏斗上过滤,用3×20毫升正己烷洗涤,滤液在旋转蒸发仪上真空浓缩。残留物在真空(<0.1mmhg,rt,30分钟)下保存,以除去己烷痕迹。产率为161.22克(77.7%)。用2×200毫升庚烷淬灭反应混合物后得到的合并水相(4升)进行萃取,然后按上述方法纯化,可分离出另外19.63克产品。
153、3-(2,3-二甲基苯基)-2-丁酮(7)
154、总产量180.85g(87.2%)。淡黄色液体。1h nmr(400mhz,cdcl3)δ7.08(d,2h),6.91(t,1h),3.97(q,1h),2.32(s,3h),2.27(s,3h),2.01(s,3h),1.35(d,3h);13с(100mhz,сdcl3)δ209.63,139.22,137.61,134.50,128.90,126.18,124.99,50.58,28.44,21.14,16.93,15.33;tr 4.88分钟(hplc面积99.2%,方法α)。
155、2,2',3,3'-四甲基联苯(9)
156、产量10.08g(7.2%)。白色晶体物质。1h nmr(400mhz,cdcl3)δ7.14(m,4h),6.96(d,2h),2.34(s,6h),1.96(s,6h);13с(100mhz,сdcl3)δ142.48,136.79,134.67,128.63,127.41,125.19,20.68,16.55;tr 42.3分钟(hplc面积98.8%,方法α)。
157、1-溴-3-(2,3-二甲基苯基)-2-丁酮(5)
158、
159、一个5l玻璃反应釜配有温控夹套,配备一个密封的机械搅拌器、一个热电偶和一个用于通入惰性气体的旋塞,将反应釜抽空(<0.1mmhg)并在90℃下干燥2小时。将反应釜冷却至室温,并充入干燥的氩气(3次泵/充气循环)。将114.57克(0.65摩尔;1.0eq)3-(2,3-二甲基苯基)丁-2-酮7与3,420毫升无水甲醇在氩气逆流下注入干燥的反应器中。将溶液冷却至-10℃,然后将124.65克(0.78摩尔;1.20eq)溴在592毫升甲醇中的溶液在搅拌(250转/分)下(30-35秒)倒入反应器(温度升至-9℃)。反应混合物的温度平稳升至-5...-6℃,并在此温度下搅拌18小时,同时用hplc(方法a)监测反应进展。接下来,反应混合物(浅橙色)的温度逐渐升高至0℃,再加入5.0克(0.03摩尔)溴,继续搅拌2.5-3小时以完成反应,同时使用hplc监测起始酮的转化率(方法a)。反应完全结束后(起始酮的转化率大于97%),将透明溶液倒入10升搅拌反应器中,反应器中装有7,200毫升去离子水,预冷至0.5℃。搅拌白色乳液30分钟,然后用4×300毫升二氯甲烷萃取。有机萃取液从反应器底部出口分离出来,合并后用1升饱和碳酸氢钠水溶液(300转/分,30分钟)和1升去离子水(300转/分,30分钟)洗涤,然后用无水硫酸钠干燥。将溶液过滤,在真空(<30℃)下除去溶剂。残留物直接用于下一步,无需进一步纯化。
160、产量171.57g(84.8%)。淡黄色透明油,无催泪性反应。1h nmr(400mhz,cdcl3)δ7.08(d,2h),6.85(t,1h),4.39(q,1h),3.79(dd,2h),2.33(s,3h),2.31(s,3h),1.40(d,3h);13с(100mhz,сdcl3)δ202.70,138.10,138.01,134.54,129.40,126.43,124.95,46.75,33.25,21.04,17.29,15.34;tr 8.16分钟(hplc面积82.0%,方法α)。
161、3-溴-3-(2,3-二甲基苯基)丁-2-酮(20)
162、
163、将80毫升乙酸乙酯中的8.0克(45.4毫摩尔;1.0eq)3-(2,3-二甲基苯基)丁-2-酮7放入100毫升玻璃反应釜中,该反应釜带有恒温夹套,配有带宽叶片的机械搅拌器和密封件,以及一个用于通入惰性气体的阀门。将溶液冷却至5℃,开始通入微弱的氩气流(100ml/min),然后在剧烈搅拌(300rpm)下,将15.25g(47.6mmol;1.05eq)吡啶氢溴酸过溴化物19以约500-600mg的份量通过塑料漏斗洒入,每隔约1分钟洒入一次。试剂的颜色立即消失,氢溴酸吡啶鎓的白色絮状结晶沉淀开始从溶液中脱落。加入全部试剂大约需要30分钟。反应混合物在5℃下搅拌40分钟,然后再加入1.5克溴化氢吡啶鎓(溴化试剂总量为16.75克)。反应混合物搅拌10分钟,然后用100毫升去离子水稀释。继续搅拌5分钟,使氢溴酸吡啶鎓沉淀溶解,排出下层水相,再用100毫升去离子水重复洗涤有机相,积极搅拌各层5分钟。水相从反应器底部出口排出,再用75毫升饱和碳酸氢钠溶液重复洗涤。各层在室温下剧烈搅拌40分钟,分离,最后用50毫升去离子水洗涤浅黄色乙酸乙酯层。水相用20毫升乙酸乙酯萃取,合并的有机萃取液在无水硫酸钠上干燥,过滤并在40℃下真空浓缩。残留物在真空中保存,以除去乙酸乙酯痕量。
164、产量12.0g(72.5%)。浅橙色油,在冷冻室(-18°с)中结晶成黄色针状物。1h nmr(400mhz,cdcl3)δ7.69(m,2h),7.16(m,1h),2.27(s,3h),2.24(s,3h),2.22(s,3h),2.09(s,3h);13с(100mhz,сdcl3)δ203.10,138.75,138.57,133.86,130.64,126.19,126.11,73.48,29.79,25.29,20.84,17.16;tr 7.58分钟(hplc面积70%,方法α)
165、3-(2,3-二甲基苯基)-2-氧代丁基乙酸酯4路线α)
166、
167、在2升圆底烧瓶中加入191.4克(1.95摩尔;3.5eq)乙酸钾和1,000毫升乙腈。将171.57克粗1-溴-3-(2,3-二甲基苯基)-2-丁酮(纯化合物5的计算含量为140.68克;0.55摩尔)在850毫升乙腈中的溶液一次加入烧瓶中,在室温(21℃)下用磁力搅拌器(1,500转/分)搅拌悬浮液,用高效液相色谱法(方法a)监测反应的进展。反应完成后(19小时,转化率大于99%),在肖特漏斗上过滤溴化钾和乙酸钾的结晶沉淀,并用2×200毫升乙腈洗涤滤器。淡黄色透明滤液在真空(<40℃)下浓缩,残余物用300毫升mtbe稀释。有机相用2×500毫升去离子水洗涤,水相再用2×50毫升mtbe萃取,合并的有机相用无水硫酸钠干燥。过滤溶液,在真空(<35℃)下除去溶剂。残留物直接用于下一步,无需进一步纯化。
168、产量158.07克(84.8%)。tr 4.54分钟(hplc面积69.3%,方法α)。
169、化合物4样品经闪蒸色谱法(硅胶60,正己烷-乙酸乙酯9:1,rf 0.16)纯化,用于核磁共振分析。纯化后的化合物4呈白色蜡状。1h nmr(400mhz,cdcl3)δ7.08(d,2h),6.91(t,1h),4.53(dd,2h),4.04(q,1h),2.31(s,3h),2.27(s,3h),2.09(s,3h),1.39(d,3h);13с(100mhz,сdcl3)δ204.59,170.26,137.86,137.80,134.34,129.27,126.36,125.13,67.08,46.93,21.10,20.48,16.73,15.30。
170、盐酸美托咪定1(方法α)
171、
172、在一个装有机械搅拌器、热电偶和高效回流冷凝器的10升玻璃夹套反应釜中,加入158.07克粗3-(2,3-二甲基苯基)-2-氧代丁基乙酸酯(纯化合物4的计算量为109.54克;0.467摩尔)到1700毫升正丙醇中的溶液。在搅拌(160转/分)下,将268.72克(1.34摩尔)一水乙酸铜(ii)在2529毫升(2293.9克;33.6摩尔)25%氨水溶液中的溶液一次加入反应器中。搅拌均匀的深蓝色溶液3分钟后,向反应器中一次加入490毫升(6.64摩尔)37%的甲酰胺。将反应混合物加热至沸腾(加热温度100℃),搅拌(160rpm)2小时。加热约10-15分钟后,开始析出甲-解托咪定3的结晶铜(i)复合物。加热2小时后,将反应悬浮液冷却至16-18℃1小时,然后在16-18°с下继续搅拌30分钟。用肖特漏斗过滤预沉淀物,并依次用1升25%的正丙醇水溶液、1升去离子水和2×250毫升丙酮洗涤。得到的深黄色精细结晶粉末在室温下真空(6毫米汞柱)干燥12小时。美托咪定铜(i)络合物的收率为99.68克(3个阶段的收率为58.1%)。
173、在一个装有机械搅拌器的10升玻璃反应釜中,加入2,850克(0.566摩尔;1.50eq)10%的二乙烯三胺五乙酸五钠盐(na5dtpa)水溶液和99.68克(0.377摩尔)美托咪定3的铜(i)复合物,悬浮在2升乙酸乙酯中。在室温(23℃)下搅拌(300-350转/分钟)24小时,直至所有固体完全溶解。深色乙酸乙酯层与浓蓝色水相分离。水相用2×200毫升乙酸乙酯萃取。将含有美托咪定碱的合并有机相转移到5升搅拌玻璃反应器中,并先后用2×500毫升0.5%na5dtpa水溶液(各30分钟)和500毫升饱和氯化钠水溶液(30分钟)洗涤。用500毫升和2×350毫升10%乙酸水溶液(搅拌速度300-400转/分,每份10分钟)依次萃取,从乙酸乙酯相中除去美托咪定。在真空中浓缩至约200毫升的乙酸乙酯层,再用2×300毫升的10%乙酸水溶液萃取美托咪定。将含有乙酸美托咪定的水提取物(1,800毫升)转移到5升搅拌玻璃反应器中,并先后用3×200毫升mtbe进行萃取,以提取少量杂质。同时,水相变亮。深色有机萃取液(mtbe)用100毫升去离子水洗涤一次,水相与含有乙酸美托咪定的萃取液合并,弃去有机相。将合并后的淡黄色水相(1,900毫升)转移到装有机械搅拌器的5升玻璃夹套反应器中,冷却至8-10℃并在搅拌下用350毫升25%的氨水溶液中和。分离出的美托咪定碱2用3×250毫升乙酸乙酯萃取,有机层用250毫升饱和氯化钠水溶液洗涤一次,用无水硫酸钠干燥,过滤,真空浓缩(注意,因为可能会起泡)。产物是一种蜂蜜状物质,呈琥珀色,在真空中会起泡,并凝固成脆弱的珍珠状泡沫。粗制美托咪定碱的产量为65.74克。
174、直接在进行浓缩的烧瓶中将美托咪定2的碱溶解在250毫升干丙酮中。将深色透明溶液冷却至-10℃,然后加入1.0克盐酸美托咪定晶体作为种子。在搅拌(500-600转/分)下,用28.0毫升36%盐酸滴加冷却溶液至ph 2-3。盐酸美托咪定1形成的沉淀开始结晶。结晶过程在-18...-20℃条件下进行23小时,产物在肖特漏斗上过滤,用3×100毫升冷(2-3℃)干丙酮洗涤,然后在40℃真空条件下干燥12小时。
175、盐酸美托咪定的收率为50.25克(56.2%,分2个阶段)。灰白色细结晶粉末(hplc面积95.3%,方法с)。残铜含量为3.7ppm(aas)。
176、纯化时,产品从含3.85%(体积分数)水的丙酮中结晶(每1克盐酸美托咪定约需9.5毫升溶剂)。产率90%(一步纯化,hplc面积98.5%,方法c)。结晶两次后,产率为78-82%(hplc面积99.5%,方法c)。重结晶后的残铜含量小于1ppm(aas)。
177、白色精细结晶粉末。1h nmr(400mhz,d6-dmso)δ9.02(s,1h),7.48(s,1h),7.04(m,2h),6.83(m,1h),4.51(q,1h),2.26(s,3h),2.24(s,3h),1.51(d,3h);13с(100mhz,d6-dmso)δ141.04,137.36,136.71,134.07,133.78,128.30,125.69,124.05,115.80,31.88,20.61,20.31,14.51;tr 3.11分钟(hplc,方法с).
178、二甲酰氨基钠21(路线β)
179、
180、将28.42克(526毫摩尔)粉末状甲醇钠和100毫升干燥甲醇注入充有氩气的500毫升圆底烧瓶中(小心,升温!),搅拌至完全溶解。在2-3分钟内将47.39克(1.052摩尔;2.0eq)甲酰胺滴加到所得溶液中。在磁力搅拌器的搅拌下,将溶液在回流温度下加热1小时。然后,稍微冷却溶液,将回流冷凝器换成带有下降冷凝器和克莱森喷嘴的蒸馏桥,克莱森喷嘴配有温度计和装有132毫升甲苯的滴液漏斗。用量筒测量馏出物的体积来蒸馏甲醇,收集约80毫升馏出物后,开始从滴液漏斗中加入甲苯,加入的速度要保持蒸馏烧瓶中溶液的体积恒定。收集蒸馏物,直到克莱森喷嘴中的蒸汽温度达到110℃(蒸馏物的总体积约为150毫升)。白色悬浮液冷却至室温,用肖特漏斗过滤。用80毫升四氢呋喃洗涤产物,并在室温下真空(0.05毫米汞柱)干燥1小时。所得产物直接用于下一步,无需进一步纯化。
181、产量45.68克(91.4%)。白色精细结晶粉末。
182、n-[3-(2,3-二甲基苯基)-2-氧代丁基]-n-甲酰甲酰胺22(路线β)
183、
184、在2l双颈圆底烧瓶中充入氩气(3个泵/充气循环),加入38.46g(404.7mmol;1.4eq)二甲酰氨基钠21和400ml乙腈(水含量<0.05%)。在磁力搅拌器上搅拌下,将87.50克粗3-(2,3-二甲基苯基)-1-溴-2-丁酮5(色谱纯度82%;281.2毫摩尔)在400毫升乙腈中的溶液一次加入到生成的悬浮液中。反应混合物在室温氩气环境下磁力搅拌20小时,然后回流2小时,使起始化合物5的转化率大于99%(hplc,方法a)。热的深黄色溶液在肖特漏斗上过滤,无机盐沉淀用50毫升乙腈洗涤。透明滤液在真空(40℃)下浓缩,得到的深黄色油用350毫升去离子水洗涤,在磁力搅拌器上搅拌10分钟。产品用300毫升二氯甲烷萃取,分离有机相并用350毫升去离子水洗涤。水相再用30毫升二氯甲烷萃取,合并的有机相在真空中浓缩。得到的黄色粘稠油直接在进行浓缩的同一烧瓶中用250毫升正己烷稀释,然后将烧瓶放入油浴(90℃)中加热,并在磁力搅拌器上搅拌30分钟。冷却混合物,小心沥干并丢弃上层正己烷,用100毫升正己烷再次洗涤剩余的油。丢弃正己烷层,在真空中去除产品中残留的正己烷。残留物直接用于下一步,无需进一步纯化。
185、产量51.24克(63.3%)。黄色粘稠油状物。1h nmr(400mhz,cdcl3)δ8.88(s,2h),7.10(m,2h),6.95(m,1h),4.30(dd,2h),4.03(q,1h),2.33(s,3h),2.26(s,3h),1.42(d,3h);13с(100mhz,сdcl3)δ209.80,163.27,138.01,137.73,134.60,129.50,126.53,125.59,48.58,46.11,21.10,16.79,15.39;tr 3.15分钟(hplc面积85.9%,方法α)。
186、1-氨基-3-(2,3-二甲基苯基)丁-2-酮盐酸盐23(路线β)
187、
188、将上一步得到的47.26克粗n-[3-(2,3-二甲基苯基)-2-氧代丁基]-n-甲酰甲酰胺22(色谱纯度85.9%;164.1毫摩尔)溶解在600毫升99%的乙醇中,然后将65.0毫升36%(764毫摩尔;4.6eq)盐酸一次性加入到得到的清黄色溶液中。在氩气环境下将溶液加热至沸腾,同时在磁力搅拌器上进行搅拌。15分钟后对反应混合物的等分试样进行hplc分析,结果显示起始化合物22向氨基酮23的转化率大于99%(hplc,方法a)。紫红色溶液在真空中浓缩,结晶残留物在40℃/2mmhg下干燥1小时(在旋转蒸发仪上)。将残留物悬浮在350毫升二乙醚中;将晶体悬浮液在磁力搅拌器上搅拌10-15分钟,在肖特漏斗上过滤,并用3×100毫升丙酮洗涤。从315毫升丙酮和30毫升去离子水的混合物中重结晶出氨基酮盐酸盐23(24.91克),并在室温下真空(6毫米汞柱)干燥12小时。
189、产量为22.0克(58.8%)。无色有光泽的针状物。1h nmr(400mhz,d6-dmso)δ8.40(s,3h),7.08(m,2h),6.88(m,1h),4.25(q,1h),3.73(dd,2h),2.26(s,3h),2.21(s,3h),1.29(d,3h);13с(100mhz,d6-dmso)δ204.41,137.95,137.22,134.53,128.83,125.92,125.00,46.37,45.45,20.66,16.58,15.01;tr 3.10分钟(hplc面积97.3%,方法β)。
190、4-[1-(2,3-二甲基苯基)乙基]-1,3-二氢-2h-咪唑-2-硫酮24(路线β)
191、
192、在250毫升圆底烧瓶中加入12.63克(55.4毫摩尔)1-氨基-3-(2,3-二甲基苯基)丁-2-酮氢氯化物23和70毫升去离子水。在磁力搅拌器的搅拌下,将26.95克(277.3毫摩尔;5.0等量)硫氰酸钾一次性加入悬浮液中,烧瓶装有回流冷凝器,反应混合物在125-130℃(浴温)下加热。加热约10分钟后,所有固体溶解,形成透明的淡黄色溶液,继续加热(约30-40分钟),产品开始析出。反应结束后(总加热时间为3小时20分钟),晶体悬浮液冷却至室温,然后降至0℃。晶体在肖特漏斗中过滤,用3×100毫升去离子水和少量96%的冷乙醇洗涤。产品在室温下真空(6mmhg)干燥12小时。
193、产量9.42克(73.1%)。淡黄色细晶光泽粉末,无味。1h nmr(400mhz,d6-dmso)δ11.83(s,1h),11.70(s,1h),7.02(m,2h),6.92(m,1h),6.50(s,1h),4.15(q,1h),2.24(s,3h),2.20(s,3h),1.38(d,3h);13с(100mhz,d6-dmso)δ141.71,136.26,133.71,133.58,133.44,127.89,125.36,123.93,111.40,31.79,20.61,19.92,14.39;tr 9.49分钟(hplc面积98.9%,方法b)。
194、美托咪定2(途径β)
195、
196、将7.90克(34.0毫摩尔)4-[1-(2,3-二甲基苯基)乙基]-1,3-二氢-2h-咪唑-2-硫酮24和50毫升99%乙醇放入500毫升的施伦克烧瓶中,烧瓶中充满氩气。在氩气的逆流中,将35毫升新制备的raney镍w-4在99%乙醇中的悬浮液(悬浮液中的镍含量为0.28克/毫升;9.8克;167毫摩尔;4.9等式)转移到烧瓶中,并在室温下用磁力搅拌器将内容物搅拌1.5小时。搅拌1.5小时后,加入第二份(35毫升;4.9当量)raney镍w-4在99%乙醇中的悬浮液,在室温下继续搅拌3.5小时。指定时间后,加入第三份(10ml;2.8g;47.7mmol;1.4eq)raneynickel w-4在99%乙醇中的悬浮液,在95°с的惰性气氛中搅拌下加热反应混合物2小时,使反应完全完成(起始化合物的转化率24≥99.5%)。黑色悬浮液冷却至室温,用肖特漏斗过滤。浅黄色透明滤液进一步通过gfa过滤器过滤,并在真空中浓缩。在室温下真空(0.05mmhg)20小时,除去乙醇痕量后,得到的淡黄色透明粘稠的美托咪定碱油结晶成多孔的轻质物。
197、产量5.85g(86%)。1h nmr(400mhz,cdcl3)δ10.56(s,1h),7.30-6.62(m,5h),4.35(q,1h),2.26(s,3h),2.17(s,3h),1.56(d,3h);13с(100mhz,cdcl3)δ143.38,141.31,136.87,134.60,134.16,128.04,125.65,124.77,117.26,34.23,21.01,20.85,14.76;tr3.12分钟(hplc面积98.0%,方法с)。
198、美托咪定2(途径c)
199、
200、称取1.29克(5.66毫摩尔)1-氨基-3-(2,3-二甲基苯基)丁-2-酮氢氯化物23和2.55克(56.6毫摩尔;10eq)甲酰胺,放入一个30毫升的玻璃瓶中。用氩气吹扫玻璃瓶,在瓶内放置一个磁性锚,盖上盖子,然后在160℃下将反应混合物加热2小时(在此之后对反应混合物的等分试样进行分析,结果显示起始化合物23的转化率大于99%)。将反应混合物冷却至室温,并用9.63毫升36%盐酸稀释。将均匀透明的黄色溶液在室温下放在磁力搅拌器上搅拌1小时,以水解过量的甲酰胺,沉淀的氯化铵悬浮液用10毫升去离子水稀释,水相用2×5毫升乙酸乙酯萃取以除去微量杂质。水相冷却至-3...-5℃,用12毫升25%的氨水溶液中和。分离出的油用2×10毫升乙酸乙酯萃取,合并的有机萃取液用10毫升去离子水洗涤,用硫酸钠干燥,过滤,真空浓缩。得到的淡黄色油状甲去甲米定碱在真空中发泡,完全去除乙酸乙酯痕迹后,结晶成乳白色块状多孔体。
201、该产品可通过从环己烷-甲苯(9:1v/v)混合物中结晶提纯,或通过上述标准程序转化为其盐酸盐。
202、产量0.43g(38%)。1h nmr(400mhz,cdcl3)δ10.56(s,1h),7.30-6.60(m,5h),4.36(q,1h),2.27(s,3h),2.18(s,3h),1.56(d,3h);13с(100mhz,cdcl3)δ143.38,141.31,136.87,134.60,134.16,128.04,125.65,124.77,117.26,34.23,21.01,20.85,14.76;tr3.12分钟(hplc a面积95.2%,方法с)。
- 还没有人留言评论。精彩留言会获得点赞!