一种单分散聚合物荧光微球的制备方法及其应用与流程
本发明涉及高分子纳米复合材料和医学检测领域,具体涉及一种聚合物荧光微球的制备方法及其在免疫侧向层析中的应用。
背景技术:
荧光微球一般指尺寸在纳米级至微米级范围内,表面或内部负载有荧光物质的微球,在外界能量刺激下能激发出荧光。由于荧光微球具有稳定的形态结构、良好的荧光特性和较高的荧光灵敏度,其在生物医学领域具有非常广泛的应用。荧光免疫层析技术利用荧光微球表面的活性官能团与抗体进行结合,可实现对血液中的待测抗原的检测。该技术通过荧光示踪可实现对检测结果的精确定量,获得的检测信号具有较高的信噪比,较高检测灵敏度和更宽的检测范围,其检测性能指标远远高于胶体金等传统快速检测技术。因此,荧光微球作为标记物的免疫层析技术具有较高的应用前景。
荧光微球的制备有物理法和化学法两种,物理法是指载体和荧光物质仅通过简单的物理作用相结合而无化学键的变化,该法制备过程简单,主要包括有吸附法、包埋法和自组装法三种,其中吸附法制备的微球由于荧光分子暴露在微球外面,容易受到外界环境及介质的影响,使检测的准确性及重现性不佳;包埋法很好地解决了物理吸附所产生的缺点,它将荧光物质完全包埋在微球内部,保障了微球荧光强度的稳定性,是目前合成荧光微球最受欢迎的方法之一。化学法则包括化学键合法和共聚法两种,这两种方法对染料和单体都有特定的要求,且化学反应有可能会改变荧光分子的结构,从而导致微球荧光性质的改变。目前,大多数技术获得的荧光微球都存在粒径分布较宽,荧光性能不稳定的缺陷。
表面带有修饰基团的荧光微球具有比表面积大、表面吸附性强、反应能力强和凝集作用大等优点,可与各种活性分子进行多种形式的反应,可固定生物酶分子,标记生物分子从而进行检测。微球表面修饰羧基或氨基的方法有后修饰法和一步法,前者是在制备成微球之后再进行官能团的修饰,该法步骤繁琐,且基团修饰量受到限制,后者是在制备微球的过程中直接引进活性官能团,该法简单有效,且活性基团含量可调,因而更有优势。
目前,关于荧光性能稳定,大小可控,粒径均一且表面官能团含量高的荧光微球制备文献报道较少,且大多数制备方法都较为繁琐。
技术实现要素:
本发明的目的是针对现有技术的不足提供一种荧光性能稳定,表面官能团含量高,大小可控及粒径均一的聚合物荧光微球的制备方法,该法采用的是细乳液聚合,操作简单,仅通过一步法即可获得微球表面含羧基或氨基的功能化荧光微球,且获得的荧光微球粒径可控,大小均一,荧光亮度高,官能团含量高,性能稳定,在免疫侧向层析中具有很好的应用前景。
本发明的第一方面提供了一种单分散聚合物荧光微球的制备方法,所述的方法包括如下步骤:
(1)提供油相混合物、水相混合物和引发剂水溶液;
(2)将所述的水相混合物与油相混合物混合,进行预乳化和完全乳化处理,从而得到备用乳液;和
(3)将所述的备用乳液和引发剂水溶液混合均匀,在氮气保护和避光条件下进行聚合反应,反应结束后进行纯化处理,从而得到聚合物荧光微球。
在另一优选例中,所述步骤3)的聚合反应在65℃~80℃下进行。
在另一优选例中,步骤3)的聚合反应在200rpm~400rpm搅拌速度下进行。
在另一优选例中,步骤3)的聚合反应时间为5~20小时。
在另一优选例中,所述的油相混合物由如下方法制备得到:提供反应单体、功能性反应单体、荧光分子和疏水剂的混合物,超声混合均匀,从而获得油相混合物;其中,
所述功能性反应单体选自下组:丙烯酸(aa)、甲基丙烯酸(maa)、丁烯酸、甲基丙烯酸-2-氨基乙酯盐酸盐(aemh)、丙烯酸二甲氨基丙酯、丙烯酸二乙氨基丙酯、丙烯酸二甲氨基乙酯、甲基丙烯酸二甲氨基丙酯、甲基丙烯酸二乙氨基丙酯、甲基丙烯酸二甲氨基乙酯;
所述反应单体选自下组:苯乙烯、甲基丙烯酸甲酯;
所述荧光分子选自下组:萘酰亚胺类染料、荧光素类染料、氟化硼二吡咯类染料、萘类染料、芴-苯并噻二唑共聚物荧光染料、硝基苯并噁二唑、钙黄绿素;二(三氯甲烷)碳酸酯。
所述的疏水剂选自下组:正十六烷、正辛醇。
在另一优选例中,所述的水相混合物由如下方法制备得到:提供表面活性剂和纯水的混合物,超声处理使表面活性剂完全溶解,从而获得水相混合物;所述表面活性剂选自下组:十二烷基硫酸钠(sds)、月硅酸钾、丁二酸二辛醋磺酸钠、十六烷基三甲基溴化铵(ctab)、十二烷基三甲基溴化铵(dtab)、十二烷基氯化铵。
在另一优选例中,所述的引发剂水溶液由如下方法制备得到:提供引发剂和纯水的混合物,振荡溶解,从而获得引发剂水溶液;所述引发剂选自下组:过硫酸钾、过硫酸胺、偶氮二异丁腈(aibn)、过氧化二苯甲酰、过氧化十二酰。
在另一优选例中,步骤2)中,所述的油相混合物和水相混合物的体积比为2:9~6:9较佳。
在另一优选例中,所述的油相混合物中的反应单体、功能性反应单体、荧光分子和疏水剂的用量比为1.8~2.0︰0~4.00︰0~0.04︰0.05~0.2。
在另一优选例中,所述的水相混合物中的表面活性剂的用量为0~0.02。
在另一优选例中,所述的聚合物荧光微球粒径为50nm~400nm,且所述的聚合物荧光微球的pdi为0~0.2。
本发明的第二方面提供了一种聚合物荧光微球,所述聚合物荧光微球的粒径50nm~400nm,pdi为0~0.2,并且所述的聚合物荧光微球是用本发明第一方面所述的方法制备。
在另一优选例中,所述的聚合物荧光微球在免疫侧向层析中作为标记物。
本发明的第三方面提供了一种制品,所述的制品含有本发明第二方面所述的聚合物荧光微球。
应理解,在本发明范围内中,本发明的上述各技术特征和在下文(如实施例)中具体描述的各技术特征之间都可以互相组合,从而构成新的或优选的技术方案。限于篇幅,在此不再一一累述。
附图说明
图1(a)为添加0.1wt%sds时微球的扫描电镜图,标尺为2.00μm。
图1(b)为添加0.5wt%sds时微球的扫描电镜图,标尺为1.00μm。
图1(c)为添加1wt%sds时微球的扫描电镜图,标尺为1.00μm。
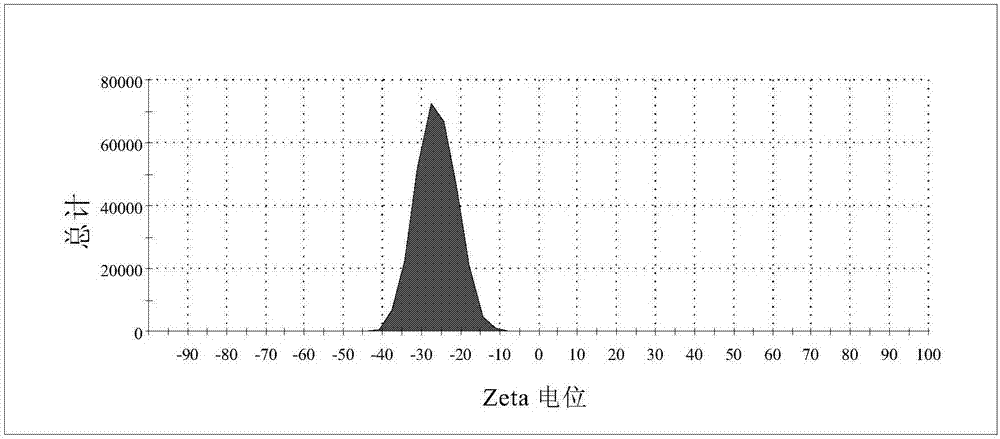
图2为带羧基基团的聚合物荧光微球的zeta电位图。
图3(a)为含有萘酰亚胺类染料的微球的荧光光谱图。
图3(b)为含有荧光素类染料的微球的荧光光谱图。
图3(c)为含有氟化硼二吡咯类染料的微球的荧光光谱图。
图3(d)为含有萘类染料的微球的荧光光谱图。
具体实施方式
为解决上述技术问题,本发明采用的一种优选的技术方案包括以下步骤:
(1)油相混合物的制备:将功能性反应单体与荧光分子、反应单体苯乙烯一起加入到超疏水剂正十六烷中,超声混合均匀,为油相混合物;
(2)水相混合物的制备:取一定量的表面活性剂分散在水中,总体积为9ml,超声处理使其完全溶解,为水相混合物;
(3)引发剂水溶液配制:将引发剂加入到1ml水中,振荡溶解;
(4)乳化过程:将水相加入到油相中,搅拌并超声进行预乳化,然后在一定功率、冰浴条件下,用细胞粉碎机处理10min至完全乳化;
(5)将上述乳液转移至100ml三口烧瓶中,加入(3)配制的引发剂水溶液,充分搅拌混合均匀,氮气保护后升温至聚合温度,在300rpm搅拌速度下避光反应5-20小时,后经透析袋透析至析出液电导率不再变化为止,以除去表面活性剂,疏水剂,盐离子和未装载的荧光分子等物质,得到纯化的聚合物荧光微球乳液。
所述荧光分子为萘酰亚胺类,荧光素类,氟化硼二吡咯类和萘类染料。
所述功能性反应单体为丙烯酸(aa)或甲基丙烯酸(maa)或甲基丙烯酸-2-氨基乙酯盐酸盐(aemh)中的一种。
所述表面活性剂为十二烷基硫酸钠(sds)或十六烷基三甲基溴化铵(ctab)类。所述引发剂为过硫酸钾或偶氮二异丁腈(aibn)或过氧化二苯甲酰中的一种。
本发明具有如下优点:
1.本发明采用经典一步法细乳液聚合来合成聚合物荧光微球,操作简单,通过改变表面活性剂的用量可调控微球粒径大小及粒径均一性;通过改变荧光分子的种类可制备不同激发和发射波长的荧光微球以满足不同仪器的要求,同时可通过改变荧光分子添加剂量来调控荧光强度。
2.本发明制备的聚合物荧光微球,粒径分布较窄,呈单一分布,具有较好的胶体稳定性。
3.本发明制备的聚合物荧光微球,由于采用的是包埋法,它将荧光分子完全包埋在微球内部,因而受外界环境影响较小,荧光性能更加稳定。
4.本发明通过改变功能性反应单体即可制备表面带有羧基或氨基基团的聚合物荧光微球,无需进一步修饰,且可通过调控功能性反应单体的剂量来调节羧基或氨基基团的含量。活性官能团可直接与生物分子进行反应,从而达到生物检测的目的。
5.本发明制备的聚合物荧光微球带有的活性官能团可与抗体进行结合,从而对待测抗原进行检测,其在生物检测行业具有较大应用前景。
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。除非另外说明,否则百分比和份数按重量计算。
实施例1:
表面活性剂添加量对聚合物微球粒径及分散性的影响
(1)油相混合物的制备:称取1.8g的苯乙烯和0.2g丙烯酸加入到0.1g超疏水剂正十六烷中,超声混合均匀,为油相混合物;
(2)水相混合物的制备:取0.002g(0.1wt%)或0.01g(0.5wt%)或0.02g(1wt%)的表面活性剂十二烷基硫酸钠(sds)分别分散在水中,总体积为9ml,振荡完全溶解,为水相混合物;
(3)引发剂水溶液配制:称取0.02g过硫酸钾加入到1ml水中,振荡溶解;
(4)乳化:将水相加入到油相中,搅拌或者超声进行预乳化,然后在250w功率,冰浴条件下,用细胞粉碎机处理10min至完全乳化;
(5)将上述乳液转移至100ml三口烧瓶中,加入上述配制的过硫酸钾水溶液,充分搅拌混合均匀,氮气保护后升温至80℃,在300rpm搅拌速度下避光反应5-20小时,得到不含染料的聚合物微球乳液;后经透析袋透析2-3天进行纯化。
采用激光粒度仪测定不同sds添加量制备的微球粒径及其粒径分布大小,结果如下:添加0.1wt%sds时制备的微球粒径为245nm,pdi(分散性指数)为0.16;添加0.5wt%sds时微球粒径为156nm,pdi为0.09;添加1wt%sds时微球粒径为96nm,pdi为0.018;通过扫描电镜测得添加0.1wt%sds、0.5wt%sds和1wt%sds时,微球粒径则分别为210nm、150nm和60nm,从pdi值和扫描电镜图均可看出微球的分散性和均一性均较好,不同sds添加量制备的微球扫描电镜图如图1所示。可见,随着表面活性剂十二烷基硫酸钠剂量的依次增大,微球粒径依次减小且粒径分布更加均匀。据此,可通过调节表面活性剂的剂量来调控微球大小及粒径均一性。
实施例2:
羧基功能化的聚合物荧光微球的制备,包括以下步骤:
(1)油相混合物的制备:称取1.8g的苯乙烯、0.2g丙烯酸和0.01g(0.5wt%)萘酰亚胺类荧光分子加入到0.1g超疏水剂正十六烷中,超声混合均匀,为油相混合物;
(2)水相混合物的制备:取0.005g的表面活性剂十二烷基硫酸钠分散在水中,总体积为9ml,振荡完全溶解,为水相混合物;
(3)引发剂水溶液配制:称取0.02g过硫酸钾加入到1ml水中,振荡使其溶解;
(4)乳化过程:将水相加入到油相中,搅拌或者超声进行预乳化,然后在250w功率,冰浴条件下,用细胞粉碎机处理10min至完全乳化;
(5)将上述乳液转移至100ml三口烧瓶中,加入上述配制的过硫酸钾水溶液,充分搅拌混合均匀,氮气保护后升温至80℃,在300rpm搅拌速度下避光反应5-20小时,后经透析袋透析2-3天除去表面活性剂,疏水剂,盐离子和未装载的荧光分子等物质,得到羧基功能化的聚合物荧光微球。
采用激光粒度仪测得微球粒径为248nm,pdi为0.019,粒径分散均一;测得微球带负电,zeta电位为-30±5mv,zeta电位图如图2所示;采用酶标仪spectramaxid3测得该荧光微球的最大激发/发射波长分别为410和491nm,如图3(a)所示。
实施例3:
氨基功能化的聚合物荧光微球的制备,包括以下步骤:
(1)油相混合物的制备:称取2.0g的苯乙烯,3.173g甲基丙烯酸-2-氨基乙酯盐酸盐(aemh),0.01g(0.5wt%)荧光素类染料加入到0.1g超疏水剂正十六烷中,超声混合均匀,为油相混合物;
(2)水相混合物的制备:取0.02g的表面活性剂十六烷基三甲基溴化铵(ctab)分散在水中,总体积为9ml,超声处理使其完全溶解,为水相混合物;
(3)引发剂水溶液配制:称取0.02g引发剂偶氮二异丁腈(aibn)加入到1ml水中,振荡溶解;
(4)乳化过程:将水相加入到油相中,搅拌或者超声进行预乳化,然后在250w功率,冰浴条件下,用细胞粉碎机处理10min至完全乳化;
(5)将上述乳液转移至100ml三口烧瓶中,加入上述配制的偶氮二异丁腈溶液,充分搅拌混合均匀,氮气保护后升温至65℃,在300rpm搅拌速度下避光反应5小时,后经透析袋透析2-3天进行纯化,除去表面活性剂,疏水剂,盐离子和未装载的荧光分子等物质,便可得到氨基功能化的聚合物荧光微球。
采用激光粒度仪测得该氨基功能化的聚合物荧光微球粒径为195nm,pdi为0.069,粒径分散均一。
实施例4:
具备不同荧光性质的聚合物荧光微球的制备,包括以下步骤:
(1)油相混合物的制备:称取1.8g的苯乙烯,0.2g丙烯酸,0.005g(0.25wt%)或0.01g(0.5wt%)或0.02g(1wt%)荧光分子加入到0.1g超疏水剂正十六烷中,超声混合均匀,为油相混合物,添加的荧光分子为荧光素类,氟化硼二吡咯类或萘类染料;
(2)水相混合物的制备:取一定量的表面活性剂十二烷基硫酸钠分散在水中,总体积为9ml,超声使其完全溶解,为水相混合物,其中含荧光素类染料的荧光微球的制备采用0.02gsds,含氟化硼二吡咯类染料的荧光微球的制备采用0.01gsds,含萘类染料的荧光微球的制备采用0.002gsds;
(3)引发剂水溶液配制:称取0.02g过硫酸钾加入到1ml水中,振荡溶解;
(4)乳化过程:将水相加入到油相中,搅拌或者超声进行预乳化,然后在250w功率,冰浴条件下,用细胞粉碎机处理10min至完全乳化;
(5)将上述乳液转移至100ml三口烧瓶中,加入上述配制的过硫酸钾水溶液,充分搅拌混合均匀,氮气保护后升温至80℃,在300rpm搅拌速度下避光反应5-20小时,后经透析袋透析2-3天进行纯化,除去表面活性剂,疏水剂,盐离子和未装载的荧光分子等物质,便可得到具备不同荧光性质的聚合物荧光微球。
采用酶标仪spectramaxid3测得含有荧光素类,氟化硼二吡咯类和萘类染料的荧光微球的最大激发/发射波长分别为482/524nm,506/520nm和420/498nm,其光谱图如图3(b)、3(c)和3(d)所示。此外,对于这三种荧光微球,随着荧光分子含量由0.25wt%逐渐增加到1wt%,微球的荧光强度均逐渐增强。将荧光微球分散在pbs以及含bsa及各种表面活性剂的缓冲液中,荧光强度没有出现降低,说明制备的微球受环境影响较小,荧光性能较好;且荧光微球乳液放置两年后,微球粒径和分散性均未见明显变化。
将含1wt%氟化硼二吡咯类染料的荧光微球与抗体进行标记,后对浓度为0、25、100、1600和5000pg/ml的相应抗原进行检测,所用免疫分析仪激发波长为468nm,接收波长为518nm,在该激发波长下,氟化硼二吡咯类染料荧光激发效率较低,在此情况下测得的荧光信号值见表1所示。由表1数据可知随着抗原浓度的增加,信号值逐渐增强,且低高值具有一定的区分度,说明该方法制备的荧光微球在免疫检测中具有潜在的应用价值。
粒径、电位与荧光性能表征
1.采用扫描电镜和激光粒度仪对聚合物微球的形貌和粒径进行分析测试,详见图1所示。结果表明:聚合物微球呈球形,分散性好,粒径均一,且大小可控。
2.采用激光粒度仪对羧基功能化的萘酰亚胺类聚合物荧光微球的zeta电位进行测试,详见图2所示。结果表明:羧基功能化的聚合物荧光微球带负电荷,电位在-26.1mv左右,表明带有羧基功能基团。
3.采用酶标仪对装有萘酰亚胺类,荧光素类,氟化硼二吡咯类和萘类染料的荧光微球进行荧光光谱扫描,如图3(a)至3(d)所示。
结果表明:含有萘酰亚胺类,荧光素类,氟化硼二吡咯类和萘类染料的荧光微球分别在最大激发波长/发射波长410/491nm,482/524nm,506/520nm和420/498nm处具有最强的荧光强度。
4.用抗体标记的氟化硼二吡咯类荧光微球对不同浓度抗原进行检测,后采用激发波长为468nm,接收波长为518nm的免疫分析仪进行信号采集,不同浓度抗原对应的信号值详见表1所示。
结果表明:随着抗原浓度的增加,信号值逐渐增强,且低高值具有一定的区分度。
表1抗体标记的含氟化硼二吡咯类染料的荧光微球对不同浓度抗原检测的信号值
在本发明提及的所有文献都在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。
- 还没有人留言评论。精彩留言会获得点赞!