一种液晶齐聚物及其制备亚5nm均孔膜的方法与流程
本发明属于新型分离材料领域,特别涉及一种液晶齐聚物及其制备亚5nm均孔膜的方法。
背景技术:
嵌段共聚物是目前制备均孔膜的主要材料。然而,由于嵌段共聚物的分子量较高(mn=50~100kg/mol),所制备的均孔膜其有效孔径较大。目前,通过嵌段共聚物制备孔径小于5nm的聚合物均孔膜存在较大挑战。此外,利用嵌段共聚物制备均孔膜,往往需借助物化手段对聚合物进行降解处理来制造孔结构。然而,对于难降解、化学惰性的嵌段共聚物,通过相应的后处置制备均孔膜显得尤为困难。因此,发展基于非嵌段共聚物的可降解新材料对于制备孔径亚5nm的均孔膜显得尤为重要。
具有光异构化性质的化合物可以通过简单地光照实现化学结构的改变或降解。邻硝基苯甲醇的光异构化特征已得到广泛研究,基于邻硝基苯甲醇的衍生物具有与邻硝基苯甲醇类似的光异构化特征,在光辐照下,邻硝基苯甲醇衍生物可以转化成相应的邻亚硝基苯甲醛,同时释放游离羧酸从而实现降解。在该专利权利要求中,我们将邻硝基苯甲醇设计到两亲性可聚合液晶小分子中,借助于两亲液晶小分子的自组装特征形成有序结构,借助两亲性液晶分子的可聚合性,在紫外辐照条件下聚合形成交联的聚合物基体,借助于邻硝基苯甲醇在紫外辐照下的可降解性实现致孔。设计合成含邻硝基苯甲醇单元的两亲性可聚合液晶分子并以此为成膜材料制备孔径小于5nm的均孔膜,是解决目前利用嵌段共聚物自组装难以制备孔径小于5nm均孔膜的有效途径。
技术实现要素:
本发明的目的是为了克服现有技术不足,提供一种液晶齐聚物及其制备亚5nm均孔膜的方法,以克服现有聚合物原料和方法难以制备出孔径亚5nm均孔膜的技术难题。
本发明采用如下技术方案:一种液晶齐聚物,分子通式如下:
其中r1,r2,r3中至少有一个是含有可聚合官能团的取代基,2位和3位中至少一个被水溶性或醇溶性聚合物取代,分别形成取代基r5、r4。
进一步地,可聚合官能团主要为烯烃、羟基、羧基、氨基、甲基丙烯酸、丙烯酸、甲基丙烯酰胺、丙烯酰胺、苯乙烯。
进一步地,取代2位的聚合物或取代3位的聚合物为分子量100~20000g/mol的聚乙二醇、聚丙二醇、聚丙烯酸、聚甲基丙烯酸羟乙酯、聚丙烯酸乙酯、聚丙烯酰胺、聚丙烯酸二甲氨基乙酯、聚丙烯酰胺二甲氨基丙胺。
一种制备亚5nm孔径均孔膜的方法,包括如下步骤:
(1)将权利要求1所述的液晶齐聚物、光引发剂a、溶剂b、添加剂c混合形成均相铸膜液;
(2)将铸膜液旋涂或浸涂在固体基底上,待溶剂自然挥发,得到初始膜;
(3)采用退火方法诱导初始膜形成组装结构;
(4)对步骤3处理后初始膜进行紫外辐照,液晶齐聚物通过可聚合的基团发生交联,形成薄膜,同时,取代基r4和r5降解;
(5)将薄膜在溶剂d中浸泡,漂洗,去除降解的取代基r4和r5,形成均孔膜。
进一步地,所述的光引发剂a选自(2,4,6-三甲基苯甲酰基)二苯基氧化膦、2-甲基苯甲酰基二苯基氧化膦、4-甲基苯甲酰基二苯基氧化膦、(2,6-二甲氧基苯甲酰基)二苯基氧化膦、4-叔丁基苯甲酰基二苯基氧化膦、4-丁基苯甲酰基二苯基氧化膦、二苯甲酮、硫杂葸酮、二苯甲酮。所述添加剂c选自氯化锂、三氟磺酸锂、三氟磺酸钠、三氟磺酸钾、六氟磷酸锂、六氟磷酸钠、六氟磷酸钾。
进一步地,所述溶剂b选自二氧六环、丙酮、氯仿、四氢呋喃、苯、甲苯、二甲基甲酰胺、二甲基乙酰胺、石油醚、乙酸乙酯中的一种或两种按照任意比组成的混合物。
进一步地,所述的固体基底选自硅片、无机玻璃、硅胶板、无纺布、聚四氟乙烯致密膜、聚乙烯亚胺致密膜以及聚砜、聚醚砜、聚偏氟乙烯、聚丙烯腈、聚乙烯、聚丙烯超滤膜、微滤膜。
进一步地,所述的紫外辐照功率为10-600w、工作波长为200-800nm、工作时间15-1200分钟。
进一步地,所述的溶剂d选自甲醇、乙醇、水、甘油中的一种或两种按照任意比组成的混合物。
进一步地,将薄膜在溶剂d中浸泡,优选的浸泡时间为0.1~24小时。
本发明与现有技术相比具有的有益效果:
(1)相对嵌段共聚物,采用液晶分子制备均孔膜的优势包括:a)液晶分子主要通过小分子之间的化学反应进行合成,结构和组成容易控制,为均孔膜的孔径调节提供了便利;b)液晶分子的合成条件相对宽松,不需要单分散性嵌段共聚物合成所需的严格低温、无水、无氧,产物容易获得;c)由于分子量较低,液晶分子取向和定向组装的速度相对较快,提高了均孔膜制备效率。
(2)本发明提供了一种利用可聚合液晶分子制备孔径小于5nm均孔膜的方法,与目前采用不具有聚合能力、分子量高、不易发生光降解的嵌段共聚物制备均孔膜的技术方案显著不同。
(3)将可聚合和可降解基团同时设计到聚合液晶小分子当中,实现了交联成膜和降解成孔的一体化,简化了成膜、成孔过程,为均孔膜的实际应用提供了便利。
(4)本方法的实施步骤简单,不需要对已有的设备进行大的改造即可实现孔径小于5nm均孔膜的量化制备。
附图说明
图1:液晶齐聚物的典型分子结构;
图2:液晶齐聚物分子tm-1的核磁图谱;
图3:液晶齐聚物分子tm-2的核磁图谱;
图4:液晶齐聚物分子tm-3的核磁图谱;
图5:交联前(a)、后(b)的红外图谱;
图6:液晶齐聚物的液晶行为及dsc图谱;
图7:液晶齐聚物成膜结构。
具体实施方式
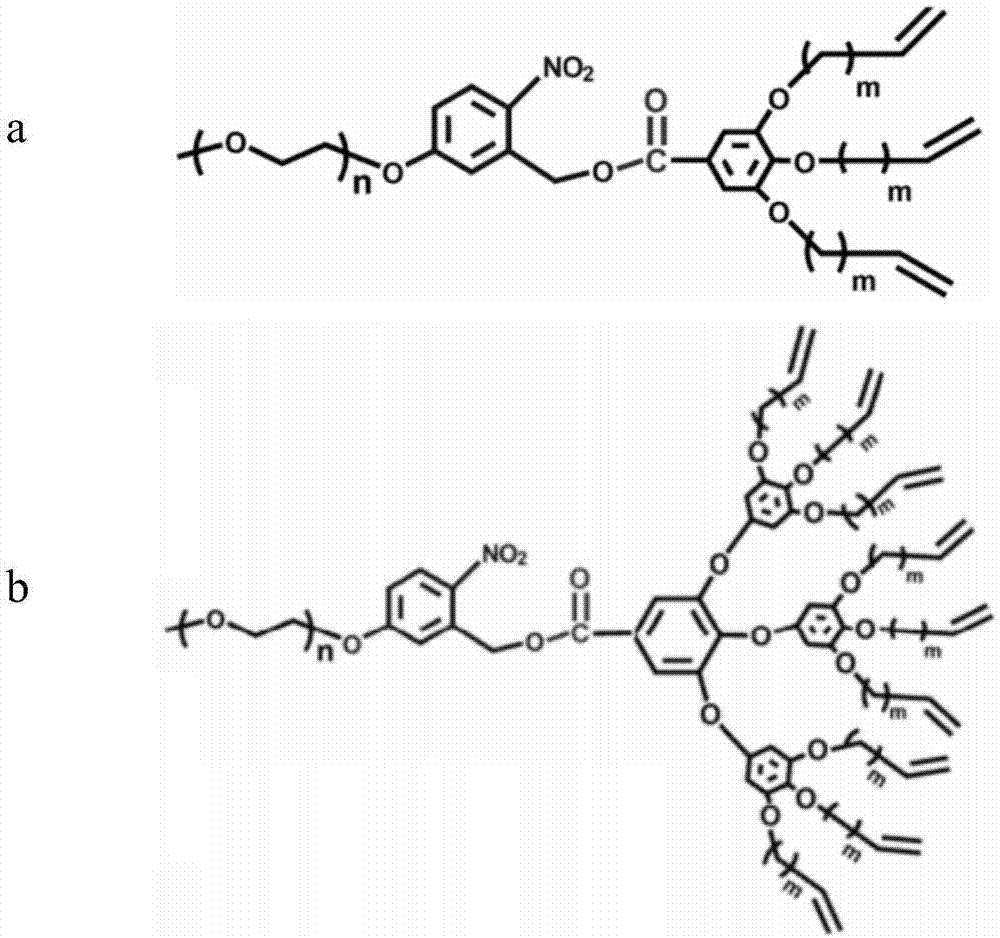
本发明采用液晶齐聚物制备亚5nm孔径均孔膜,液晶齐聚物含有在紫外辐照下可降解的邻硝基苯甲酯官能团及衍生结构,其分子通式如下:
其中r1,r2,r3中至少有一个是含有可聚合官能团的取代基,可在紫外辐照下发生聚合,使得液晶齐聚物发生交联,形成薄膜,r1,r2,r3为含有可聚合官能团的取代基,其中r1,r2,r3的结构可以相同,也可以有所差异,可以是线性结构,也可以是支化结构。
上述结构中,2位被水溶性或醇溶性聚合物取代,形成取代基r4,3位被水溶性或醇溶性聚合物取代,形成取代基r5,取代基r4和r5分子量不同,一方面不同的分子量大小有利于形成的组装尺度不同,另一方面,水溶性或醇溶性有利于降解被漂洗。
本发明液晶齐聚物的典型结构式如图1a和b所示。
通过本发明,获得的均孔膜能达到的最大孔径小于5nm。
下面结合具体实施例对本发明作进一步说明。
实施例1
一种用于制备孔径2.5nm左右均孔膜的液晶齐聚物的合成方法:
1、将二乙二醇甲醚(10g,83.23mmol)、三乙胺(16.84g,166.46mmol)加入到二氯甲烷(100ml)中,搅拌均匀,混合物冷却至0℃。再将4-甲基苯磺酰氯(31.74g,166.46mmol)的二氯甲烷溶液(50ml)滴加到混合物中,室温下搅拌反应48h。反应结束,将反应液冷却到室温,反应液用1mol/l盐酸洗涤(2×200ml),再用饱和盐水洗涤(2×200ml),无水硫酸镁干燥,减压蒸去溶剂,经过柱层析分离得到产物(物质a-1),收率:79.5%。
2、将物质a-1(3g,10.93mmol)、5-羟基-2-硝基苯甲醇(1.85g,10.93mmol)、碳酸钾(3.02g,21.86mmol)加入到丙酮(150ml)中,搅拌均匀,反应液室温通氮气30min。氮气保护下,65℃搅拌反应48h。反应结束,将反应液冷却到室温,然后过滤,滤液减压蒸去溶剂,残留物加硅胶制砂,经过柱层析,得到淡黄色晶体(物质a-2),收率:11.4%。
3、将没食子酸甲酯(3.33g,18.1mmol)、碳酸钾(25g,181mmol)加入到dmf(80ml)中,搅拌均匀,混合物冷却至0℃,通氮气30min。再将11-溴-1-十一醇(15g,60mmol)滴加到混合物中。氮气保护下,75℃搅拌反应20h。反应结束,将反应液冷却到室温,然后过滤,滤液减压蒸去溶剂,残留物溶于去离子水(80ml),趁热用乙酸乙酯(2×70ml)萃取,合并有机层,再用无水硫酸镁进行干燥。减压蒸去溶剂,得到白色固体(物质a-3)。收率:69.5%。
4、将物质a-3、氢氧化钾(5.61g,0.1mol)加入到3:1的甲醇/水的混合溶液(100ml)中,搅拌至完全溶解。60℃搅拌反应12h。反应结束,用3mol/l的盐酸中和至中性。趁热用乙酸乙酯(2×70ml)萃取,合并有机层,再用无水硫酸镁进行干燥。减压蒸去溶剂,得到白色固体(物质a-4)。收率:86.6%。
5、将物质a-4(8g,11.8mmol)、n,n-二甲基苯胺(5.35g,44.1mmol)、2,6-二叔丁基对甲酚(150mg)加入到1,4-二氧六环(65ml)中,搅拌至完全溶解,再将丙烯酰氯(4g,44.1mmol)滴加到混合物中。60℃搅拌反应2.5h。反应结束,将反应液冷却到室温,滴加甲醇(1ml)淬灭,减压蒸去溶剂,得到白色固体,加1mol/l盐酸(300ml)搅拌,过滤,固体(物质a-5)冷冻干燥2天。收率:97.8%。
6、将物质a-2(0.71g,2.62mmol)、物质a-5(2.53g,3mmol)、4-二甲氨基吡啶(100mg,0.82mmol)和二环已基碳二亚胺(842mg,4mmol)加入到二氯甲烷(30ml)中。室温搅拌反应48h。反应结束,过滤,滤液减压蒸去溶剂,再加乙酸乙酯(20ml)溶解,再过滤,反复2次,除杂质dcu。最后,经过柱层析,得到目标产物(tm1),收率:43.4%。上述结构经过核磁共振光谱仪分析,其结果如核磁图谱如图2所示。可以看出,上述合成方法可以实现目标产物的合成,结构式如下:
实施例2
一种用于制备孔径3.5nm左右均孔膜的液晶齐聚物的合成方法:
1、将聚乙二醇550单甲醚(45.78g,83.23mmol)、三乙胺(16.84g,166.46mmol)加入到二氯甲烷(100ml)中,搅拌均匀,混合物冷却至0℃。再将4-甲基苯磺酰氯(31.74g,166.46mmol)的二氯甲烷溶液(50ml)滴加到混合物中,室温下搅拌反应48h。反应结束,将反应液冷却到室温,反应液用1mol/l盐酸洗涤(2×200ml),再用饱和盐水洗涤(2×200ml),无水硫酸镁干燥,减压蒸去溶剂,经过柱层析,得到油状产物(物质b-1),收率:74.7%。
2、将物质b-1(7.70g,10.93mmol)、5-羟基-2-硝基苯甲醇(1.85g,10.93mmol)、碳酸钾(3.02g,21.86mmol)加入到丙酮(150ml)中,搅拌均匀,反应液室温通氮气30min。氮气保护下,65℃搅拌反应48h。反应结束,将反应液冷却到室温,然后过滤,滤液减压蒸去溶剂,残留物加硅胶制砂,经过柱层析,得到淡黄色液体(物质b-2),收率:54.2%。
3、将没食子酸甲酯(3.33g,18.1mmol)、碳酸钾(25g,181mmol)加入到dmf(80ml)中,搅拌均匀,混合物冷却至0℃,通氮气30min。再将11-溴-1-十一醇(15g,60mmol)滴加到混合物中。氮气保护下,75℃搅拌反应20h。反应结束,将反应液冷却到室温,然后过滤,滤液减压蒸去溶剂,残留物溶于去离子水(80ml),趁热用乙酸乙酯(2×70ml)萃取,合并有机层,再用无水硫酸镁进行干燥。减压蒸去溶剂,得到白色固体(物质b-3)。收率:69.5%。
4、将物质a-3、氢氧化钾(5.61g,0.1mol)加入到3:1的甲醇/水的混合溶液(100ml)中,搅拌至完全溶解。60℃搅拌反应12h。反应结束,用3mol/l的盐酸中和至中性。趁热用乙酸乙酯(2×70ml)萃取,合并有机层,再用无水硫酸镁进行干燥。减压蒸去溶剂,得到白色固体(物质b-4)。收率:86.6%。
5、将物质b-4(8g,11.8mmol)、n,n-二甲基苯胺(5.35g,44.1mmol)、2,6-二叔丁基对甲酚(150mg)加入到1,4-二氧六环(65ml)中,搅拌至完全溶解,再将丙烯酰氯(4g,44.1mmol)滴加到混合物中。60℃搅拌反应2.5h。反应结束,将反应液冷却到室温,滴加甲醇(1ml)淬灭,减压蒸去溶剂,得到白色固体,加1mol/l盐酸(300ml)搅拌,过滤,固体(物质b-5)冷冻干燥2天。收率:97.8%。
6、将物质b-2(1.84g,2.62mmol)、物质b-5(2.53g,3mmol)、4-二甲氨基吡啶(100mg,0.82mmol)和二环已基碳二亚胺(842mg,4mmol)加入到二氯甲烷(30ml)中。室温搅拌反应48h。反应结束,过滤,滤液减压蒸去溶剂,再加乙酸乙酯(20ml)溶解,再过滤,反复2次,除杂质dcu。最后,经过柱层析,得到目标产物(tm2),收率:68.0%。上述结构经过核磁共振光谱仪分析,其结果如核磁图谱如图3所示。可以看出,上述合成方法可以实现目标产物的合成,结构式如下:
图6是实施例1和实施例2所制备的液晶齐聚物的液晶行为及dsc图谱。图6显示,光可降解模块的引入使得液晶分子的液晶属性消失。然而,通过添加添加剂可以使得液晶属性恢复,同时可以有效的调节液晶属性的温度区间,为后期退火过程提供便利。
实施例3
一种用于制备孔径5nm左右均孔膜的液晶齐聚物的合成方法:
1、将聚乙二醇1000单甲醚(83.23g,83.23mmol)、三乙胺(16.84g,166.46mmol)加入到二氯甲烷(200ml)中,搅拌均匀,混合物冷却至0℃。再将4-甲基苯磺酰氯(31.74g,166.46mmol)的二氯甲烷溶液(50ml)滴加到混合物中,室温下搅拌反应48h。反应结束,将反应液冷却到室温,反应液用1mol/l盐酸洗涤(2×200ml),再用饱和盐水洗涤(2×200ml),无水硫酸镁干燥,减压蒸去溶剂,残留物加硅胶制砂,经过柱层析,得到蜡状产物(物质c-1),收率:60.8%。
2、将物质c-1(12.62g,10.93mmol)、5-羟基-2-硝基苯甲醇(1.85g,10.93mmol)、碳酸钾(3.02g,21.86mmol)加入到丙酮(150ml)中,搅拌均匀,反应液室温通氮气30min。氮气保护下,65℃搅拌反应48h。反应结束,将反应液冷却到室温,然后过滤,滤液减压蒸去溶剂,残留物加硅胶制砂,经过柱层析,得到淡黄色液体(物质c-2),收率:70.2%。
3、将没食子酸甲酯(3.33g,18.1mmol)、碳酸钾(25g,181mmol)加入到dmf(80ml)中,搅拌均匀,混合物冷却至0℃,通氮气30min。再将11-溴-1-十一醇(15g,60mmol)滴加到混合物中。氮气保护下,75℃搅拌反应20h。反应结束,将反应液冷却到室温,然后过滤,滤液减压蒸去溶剂,残留物溶于去离子水(80ml),趁热用乙酸乙酯(2×70ml)萃取,合并有机层,再用无水硫酸镁进行干燥。减压蒸去溶剂,得到白色固体(物质c-3)。收率:69.5%。
4、将物质c-3、氢氧化钾(5.61g,0.1mol)加入到3:1的甲醇/水的混合溶液(100ml)中,搅拌至完全溶解。60℃搅拌反应12h。反应结束,用3mol/l的盐酸中和至中性。趁热用乙酸乙酯(2×70ml)萃取,合并有机层,再用无水硫酸镁进行干燥。减压蒸去溶剂,得到白色固体(物质c-4)。收率:86.6%。
5、将物质c-4(8g,11.8mmol)、n,n-二甲基苯胺(5.35g,44.1mmol)、2,6-二叔丁基对甲酚(150mg)加入到1,4-二氧六环(65ml)中,搅拌至完全溶解,再将丙烯酰氯(4g,44.1mmol)滴加到混合物中。60℃搅拌反应2.5h。反应结束,将反应液冷却到室温,滴加甲醇(1ml)淬灭,减压蒸去溶剂,得到白色固体,加1mol/l盐酸(300ml)搅拌,过滤,固体(物质c-5)冷冻干燥2天。收率:97.8%。
6、将物质c-2(3.01g,2.62mmol)、物质c-5(2.53g,3mmol)、4-二甲氨基吡啶(100mg,0.82mmol)和二环已基碳二亚胺(842mg,4mmol)加入到二氯甲烷(30ml)中。室温搅拌反应48h。反应结束,过滤,滤液减压蒸去溶剂,再加乙酸乙酯(20ml)溶解,再过滤,反复2次,除杂质dcu。最后,经过柱层析,得到目标产物(tm3),收率:55.8%。上述结构经过核磁共振光谱仪分析,其结果如核磁图谱如图4所示。可以看出,上述合成方法可以实现目标产物的合成,结构式如下:
实施例4
一种制备厚度为100nm孔径为2.5nm左右均孔膜的方法:
(1)将tm1,光引发剂(2,4,6-三甲基苯甲酰基)二苯基氧化膦、添加剂三氟磺酸锂溶解在溶剂中形成均相溶液,其中tm1、(2,4,6-三甲基苯甲酰基)二苯基氧化膦、三氟磺酸锂的质量浓度分别为2.5wt%,0.1wt%,0.25wt%;
(2)将配置好的溶液旋涂在硅片上,旋涂条件为500r/min保持10s,再3000r/min保持30s;
(3)将旋涂好的样品用pdms板覆盖并用重物压实,放入程序升温烘箱内进行退火,升温速率5℃/min,降温速率0.1℃/min;
(4)将样品置于紫外光下照射10h,进行交联以及成孔;光照前后样品的红外图谱如图5所示,证明液晶齐聚物通过可聚合的基团发生交联,形成薄膜,同时,取代基r4和r5降解;
(5)将样品用清水浸泡洗涤,以除去降解的模块,通过电镜观测,所制备的膜孔径约为3.5nm,其电镜结果如图7所示。由于膜厚度较薄同时膜孔径小,所制备得到的膜呈透明状。同时,膜本身具有一定的强度,证明紫外交联确实有效的发生了。
膜厚度可以进一步通过改变溶液浓度和旋涂条件进行调整。
实施例5
一种制备厚度为200nm孔径为5nm左右均孔膜的方法:
(1)将tm3,光引发剂二苯甲酮、添加剂氯化锂溶解在溶剂中形成均相溶液,其中tm3、二苯甲酮、氯化锂的质量浓度分别为5wt%,0.2wt%,0.5wt%;
(2)将洁净的硅片浸没到配置好的溶液中,小心取出,沥去多于溶液;
(3)将制备好的样品用pdms板覆盖并用重物压实,放入程序升温烘箱内进行退火,升温速率5℃/min,降温速率0.1℃/min;
(4)将样品置于紫外光下照射20h,进行交联以及成孔;
(5)将样品用清水浸泡洗涤,以除去降解的模块,通过电镜观测,所制备的膜孔径约为4nm。
膜厚度可以进一步通过改变溶液浓度进行调整。
- 还没有人留言评论。精彩留言会获得点赞!